1997諾貝爾醫學獎
Prion粒子–––新型感染源的發現
Prion的歷史
歐洲十八世紀,就已經有Scrapie(羊搔癢症)的紀錄。這些生病的羊隻會有行動不穩、無法站立、顫抖、因劇烈的搔癢而造成羊隻磨蹭樹幹以至於刮去(scrape off)身上的毛、最終導致羊隻死亡。
二十世紀前葉,庫茲菲德醫師和雅各醫師分別發現有種疾病,病人的腦部嚴重受損卻沒有發炎的情況。而這種病就被命名為Creutzfeldt-Jakob disease(庫賈氏症)。
1950年代中期,新幾內亞的富雷(Fore)部落爆發了Kuru(枯魯症)。
Kuru在當地語言是顫抖之意,發病的多屬當地的婦女與小孩。當地三萬五千左右的人口,在當時已有三千人左右死於Kuru症。Carleton Gajdusek(加德賽克)醫師與Vincent Zigas(吉加斯)醫師,進入當地部落研究,在解剖患者死亡後的屍體腦部組織後發現腦部受損嚴重,有許多海綿狀的小孔,腦部沒有發炎的症狀。
1996年,英國爆發狂牛症(BSE, Bovine Spongiform Encephalopathy)。在發病的牛身上也可以發現腦組織海綿狀、運動不協調、平衡困難等等與庫賈氏症相似的痕跡。
1976年獲得諾貝爾醫學獎的Gajdusek醫師,在發現Kuru症後做了實驗,把由Kuru症患者腦部的萃取液打入黑猩猩的腦部,結果黑猩猩出現十分相似的症狀。當時已發現具有感染力的萃取液在經過加熱、紫外線照射等常規的消毒發法皆無法使萃取液喪失感染力。
1997年獲得同一獎項的Stanley B. Prusiner則提出了傳染性蛋白質(Prion)為這一系列疾病的致病體,經過實驗後更進一步發現,製造prion蛋白質的基因本來就存在於人體的染色體,還有許多動物的染色體中。造成prion蛋白致命的因素在於蛋白質的立體結構,而非胺基酸序列上的錯誤所導致。正常PrP,又稱PrPc ;傳染性PrP(PrPres),兩者的差別在於立體結構中PrPc以αhelix為主,而PrPres以βsheet較多。
由於PrPres在溶體內產生澱粉狀堆積,最後使溶體撐破、細胞分解。
在細胞死亡後原先存在與細胞內的繼續攻擊鄰近的細胞,最後造成腦部的海綿狀現象,影響個體中樞系統運作,造成死亡。
Prion 致病理論的先驅,Stanley B. Prusiner 的研究小組於2004年七月刊登於 Science 期刊的論文「Synthetic Mammalian Prions」中指出,若使用基因轉植的技術利用大腸桿菌來表現 prion 蛋白(Recombinant mouse prion protein, recMoPrP),並將此蛋白質團塊植入小鼠腦中,則可以在小鼠腦中發現異變型的 PrP 堆積,且這些異變型 PrP 還可以再用來感染其他小鼠。
在Cell雜誌2005年四月二十二出版的內容中,標題為「In Vitro Generation of Infections Scrapie Prions」,使用protein misfolding cyclic amplification, PMCA技術,,用以證明是PrPres而非PrPC帶有感染力。PMCA技術是將取自患病倉鼠腦中的異變型蛋白PrPres 與來自一般倉鼠的正常型蛋白PrPC 相混合,待PrPres 誘使 PrPC 聚集之後,再將之以超音波打散,接著再從前述步驟的產物中取出一部分加入更多的 PrPC予以稀釋。如此重複直至原始的 PrPres種子被稀釋至十的負十或負二十次方。
根據過去的研究,經過這種程度稀釋的 PrPres 已經無法顯現出感染力。
然而這些 PMCA 產物仍然被觀察到具有感染力,暗示原本為正常型的 PrPC 的確被轉換成了具有感染力的成分。
在二十世紀末歐洲爆發的狂牛症,以及時間相差不久的變異性庫賈氏症被懷疑彼此間有關聯性。可能是由於食用了帶有PrPres的牛肉,過了數年後發病。而狂牛症的可能由於餵食牛隻的飼料裡摻有牛隻、羊搔癢症的羊做成的骨粉,而吸收了prion蛋白。原先飼料摻骨粉的目的在於讓牛直接吸收蛋白質,讓牛長得更壯。結果造成BSE爆發的疫情。
有另一種懷疑,是由於同類互食的結果。牛本來是草食性動物,在吃了肉做的飼料以後出現狂牛症;分食屍體的食腦行為,導致新幾內亞的枯魯症。尼安德塔人的骨頭上曾被發現有被屠宰過的痕跡,考古學家也曾在十二世紀的美洲人糞便和鍋子裡發現人類的肌肉蛋白質。遠古的行為,也許造成基因上的變異,成為prion疾病的起源。
Prion疾病病理
|
Scrapie ( 羊 ) |
1732 |
|
CJD ( 人 ) |
1920 |
|
GSS ( 人 ) |
1936 |
|
Kuru disease ( 人 ) |
1957 |
|
Prion蛋白 |
1982 |
|
FFI ( 人 ) |
1986 |
|
MCD ( 牛 ) |
1986 |
|
vCJD ( 人 ) |
1996 |
|
CWD ( 鹿 ) |
罕見 |
|
TME ( 水貂 ) |
罕見 |
|
|
|
上表為各疾病及Priom所被發現的年代,以表示歷史上的先後順序。
CJD全名為Creutzfeldt-Jakob disease,又稱Transmissible Spongiform Encephalopathy,TSE,傳播性海綿樣腦症。最大的特徵為病理解剖上有大腦皮質海綿狀空洞,在全球年發生率約為百萬分之0.5~1,雖然已屬罕見,但為人類身上最多案例的Prion疾病。
在台灣1997~2003年中發生134例,年發生率為百萬分之0.36,和全球發生率相比還蠻接近。以發生在55~75歲的中老年人為主,平均發病年齡65歲。以發病原因來說分成3型。散發型約佔85~90﹪,和突變有關。家族遺傳型約佔5~10﹪。醫原污染型比例少於5﹪,最著名案例為美國70年代的腦下腺素注射計畫,雖然效果良好,使許多侏儒長高,但也使其中一些侏儒染上CJD而死亡。
初期病徵有記憶力衰退、行為異常、步態不穩、肌躍症,後期則嚴重痴呆(不說話也不會動)終至死亡。發病後大多在一年內死亡,,平均為4個半月死亡。臨床上可以腦電圖、腦脊髓液、核磁共振檢查腦,電圖中有短間隔陣發性棘波為特徵但,但臨床診斷的正確率約為80%,所以通常病人死亡後仍需作病理解剖來作確認。
|
|
|
|
|
|
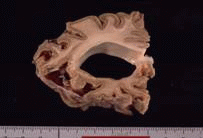
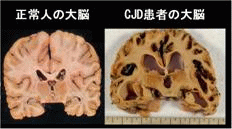
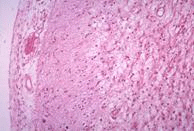
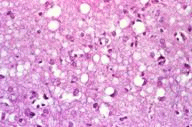
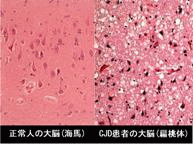
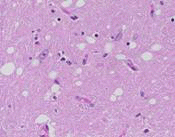
| 患者的腦部,可看出有大腦萎縮的現象。 | 患者的腦部和正常人腦部比較圖,可看出有大腦萎縮的現象。 | 患者的腦部皮質病理切片,有許多海綿狀空泡。 | 患者的腦部皮質病理切片,有許多海綿狀空泡。 | 腦部皮質病理切片,和正常人作比較後,可看出在細胞間質中有許多海綿狀空泡。 |
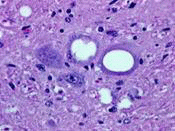
vCJD全名為variant CJD,被認為和狂牛症高度相關,全球至2006年為止 約有180個病例,其中約160個病例在英國,超過90﹪以上病歷在英國,不過在台灣尚未發現此種病症。發病範圍在12~52歲,其中以年輕人為主,平均發病年齡為29歲,初期病徵有憂鬱症、肌肉不協調,但是少肌躍症,後期則為嚴重痴呆直至死亡發病後平均14個月死亡。臨床上可作腦電圖(無短間隔陣發性棘波),核磁共振檢查來辦別,但病人死後仍需作病理解剖來進一部判定,病理切片特徵有類澱粉樣蛋白沉澱斑塊+周圍有海綿樣空泡。
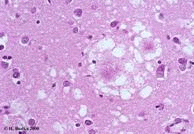 |
右圖為vCJD患者的腦部皮質病理切片, 可看出類澱粉樣蛋白沉澱斑塊+周圍有海綿樣空泡。
|
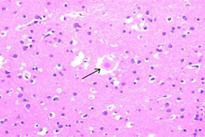 |
右圖為vCJD患者的腦部皮質病理切片, 可看出類澱粉樣蛋白沉澱斑塊+周圍有海綿樣空泡。
|
Kuru disease 為一地區性疾病,發生在巴布亞新幾內亞東部Fore族,Kuru為Fore族語的顫抖之意。此土著有吃人肉的習俗,當一個人死去時,其親友會分食其肉體,男人只吃少量人類肌肉,大多吃豬肉,女人及小孩則只靠吃人肉、人腦來補充蛋白,因而Kuru disease常發生在女人和小孩身上。病徵有全身顫抖、肌肉不協調、似笑非笑、痴呆直至死亡,發病後6~9個月死亡。病理解剖上特徵有澱粉樣蛋白沉澱斑塊及海綿空泡。當病因發現後澳洲政府就禁止吃人肉習俗,kuru症就戲劇性低消失。
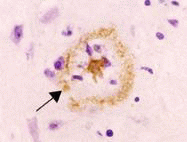
右圖為Kuru disease患者的腦部皮質病理切片,
可看出在不同染色下的類澱粉樣蛋白沉澱斑塊。
GSS全名為Gerstmann-Straussler-Scheinker Syndrome,在澳洲首先被發現,為家族性顯性遺傳疾病,此疾病十分罕見,發生率十億分之20~50,至目前為止只證實五十多個家族病歷。此疾病只發生在成年人身上,病徵類似CJD,發病後可活2個月~12年,通常在二~六年間死亡。
FFI 全名為Fatal Familial Insomnia,致死性家族失眠症,為家族性遺傳疾病,較GSS更為罕見,全球目前只證實六個家族病歷。此疾病常發生在成年人身上,初期病徵有難治性失眠、腦神經功能紊亂和運動障礙,後期則昏迷值至死亡,病理解剖特徵有丘腦萎縮。
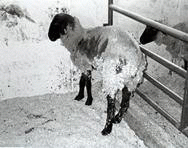
Scrapie,羊騷症,在18世紀就已在歐洲多國大量被發現,但是對人類沒有危害,所以並不受重視。病徵有步態失衡、顫抖、以身體刮擦欄杆來搔癢,病理解剖可發現大腦皮質有海棉樣空泡。
下圖為患Scrapie的羊腦部皮質病理切片,有許多海綿狀空泡。
BSE 全名為Bovine Spongiform Encephalopathy,又稱Mad Cow Disease,MCD,在BSE被發現之前,就長期以羊骨粉作高蛋白飼料,當患有Scrapie的羊作成羊骨粉來餵牛,則使牛感染Prion而得到BSE。
Prion從羊感染至牛為自然界Prion第一次打破物種壁障,
從牛感染到人為第二次打破物種壁障。
至2003年11月為止,英國有183,616個病例,而其他國家有4,608個病例,超過90﹪以上病歷在英國,病徵有神經質、具攻擊性、走路不穩、肌躍,發病後2星期~6個月死亡。
右圖為患BSE的羊腦部皮質病理切片,有許多海綿狀空泡。
從這些疾病看來,海綿狀空泡為Prion所引起的疾病的共同特徵。
CWD 全名為Wasting disease,慢性損耗疾病,為發生在鹿身上的疾病,只發生在北美洲。TME 全名為Transmissible Mink Encephalopathy,傳染性貂腦症,是發生在水貂身上的疾病,只發生在北美洲。這兩種疾病皆十分罕見。
Prion結構分析
Prion 電泳,為Stanley B. Prusiner博士研究prion所做的實驗之一,使用的方法為:聚丙烯醯胺膠體電泳PAGE(polyacrylamide gel electrophoresis)。
蛋白質為胺基酸組成,其帶電情形受擁有的胺基酸影響,如Asn(asparagine)、Gln(glutamine)在支鏈上有氨基”-NH3” ,水中解離後帶正電;Asp(aspartic asid) 、Glu(glutamic acid)在支鏈上有”-COOH”基,解離後帶負電。另外有些胺基酸的支鏈根本不帶電。而且,在蛋白質分子未呈現鏈狀時,電泳時會影響其速度,使之難以由做為長度對照的maker相比較。
在這種情況下,直接拿去電泳的結果,對想知道胺基酸序列長短沒有幫助。
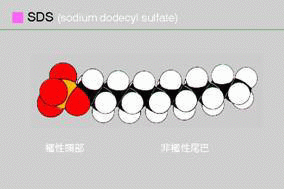
因此,蛋白質電泳時會加入一界面活性劑:SDS。
SDS具一親油端、一親水端,親油端和蛋白質的backbone相連結,
使帶負電的SDS親水端朝外。
SDS-PAGE 系統中,除了整個電泳系統含有 0.1% SDS
外,樣本也要加入 SDS (並以 mercaptoethanol
打斷雙硫鍵) 同時加熱處理。
則蛋白質會解構成為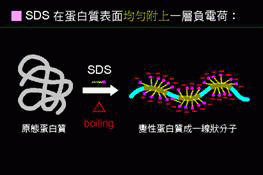
一條直鏈狀分子,其上並佈滿了 SDS 的負電荷;
請注意,理論上 SDS 是很均勻的吸附到蛋白質上,
因此不管原來蛋白質分子的大小,每種蛋白質分子
上所吸附的負電荷密度是相同的。
如此一來,就可以和已知長度,且上面也
吸附同密度負電荷的marker,做長度上的判別。

1號2號為未感染的倉鼠prion (prpc);3號4號為
被感染的倉鼠prion (prpc,prpsc)。
在2號和4號,施以proteinase K。充分反應後,
prpc完全水解,2號沒有蛋白質存在;4號留下
分子量較小的蛋白,稱作prp 27~30(kd),這是N
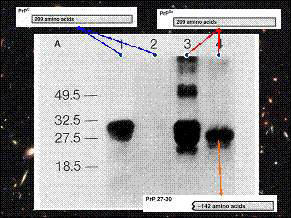 端被水解約67個胺基酸後留下的蛋白質。
端被水解約67個胺基酸後留下的蛋白質。
顯示prp27~30是prpsc的抵抗水解脢核心。
可是prpc、prpsc同樣具有這段序列,為何僅prpsc
會留下prp27~30,而prpc則否?仍產生科學家間的爭議。
3號電泳結果,發現prion的分子量不一致,而且部份分子量是原prion的2、3倍,可以和「prpsc使prpc同化後,聚集成類似晶體的形式」相呼應。
prion序列為254個codon所轉錄出,經轉錄後調控,N端C端各去掉一段序列,得到209個胺基酸的prpc、prpsc蛋白,兩者在胺基酸數量上並無
差異。
此三圖為電子顯微鏡下的prpc、prpsc、prp27~30。prp、prpsc的聚集情形,都沒有出現可辨識的多聚體;而prp27~30卻出現明顯桿狀的聚合物,和澱粉體幾乎相同(澱粉體為阿茲海默症患者腦部內的病徵)。不禁引起臆測:
「prp27~30,這個prpsc水解後留下的抗水解片段,是否為後來導致各種prion疾病的元兇?」
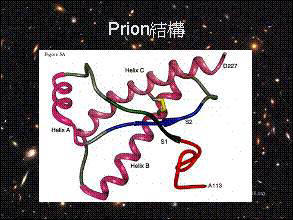
正常倉鼠prion (prpc) 的 prp27~30 序列 3D模型。粉紅色的helixA(144-157)、helix(172-193)、helixC(200-227)為α-helix的結構;
紅色的胺基酸序列(113-126),形成一個親油性區;黑色箭頭(129-131)S1、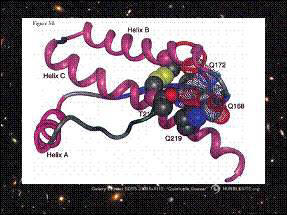
藍色箭頭(161-163)S2,兩條反向形成β-pleated sheat;
Cys179、Cys214構成雙琉鍵,使helixB、
helixC連接,在異常prion(prpsc)中,
也穩定存在。
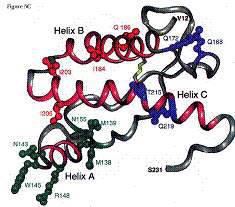
Protein X 是一種在prpsc存在時,將催化prpc構形改變為prpsc,類似chaperone功能的蛋白質。由於凡得瓦力和靜電力的影響,protein X將嵌入helixB的N端、helixC的中間表面。protein X 的參與,改變了 prpc的3D結構。
以紅色標記的184、186、203、205胺基酸,推測是抗原
決定基(epitope)的位置;綠色標記的138、139、143、145、
148、155胺基酸,可能操控prion在物種間傳遞的能力;
168、172、215、219則是和protein X的相連位置

此圖為prion蛋白的活性大小位置偵測結果。
上方以藍色為主的是prion蛋白,下方紅色長鏈是額外加進去的胺基酸序列,
作為對照。紅色(負值)表示活性大,容易形變;
藍色(正值)表示活性小。顯示prion在平常情況趨於穩定,
不是那麼容易就可以發生形變。
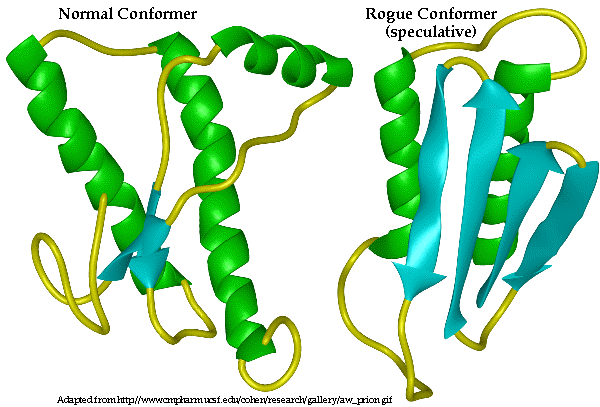
圖為人類prpsc的三級結構。108-113、116-122形成折板結構;
128-135、138-144也形成折板;以棒撞球狀顯示的結構,可能是prion的種族屏障原因。
PrPc主要結構是α-helix,幾乎沒有β-sheet;而改變後的PrPsc結構卻已β-sheet為主。
將感染小鼠的PrP去感染將PrP基因剔除(gene knockout)的小鼠則發現該小鼠不會發病,顯示PrPc是使PrPsc增加的原料。
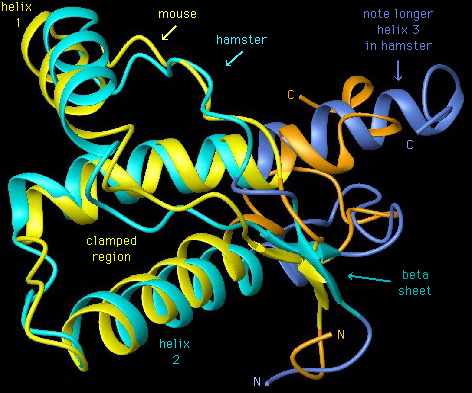
將感染倉鼠的PrPsc去感染小鼠,結果發現小鼠不會發病。但將此PrPsc去感染有倉鼠PrP基因轉植的小鼠,結果發現小鼠發病了。此實驗提供了誘發假說的證據,也說明了造成種族屏障的原因是因PrP的基因胺基酸序列不同。如倉鼠和小鼠的PrP基因有16個胺基酸不同。
若PrP基因序列越相近,則跨越種族的感染發生的機率也越高。羊與牛的PrP基因差異很小,所以感染羊的PrPsc很容易就會感染牛隻;而牛與人類的PrP基因序列有相當大的差異,所以PrPsc從牛傳染給人的機率是相對較低的。
致病的機制有二:
1. 因基因發生突變而產生了不正常的PrP,不正常的PrP在神經細胞的溶體內堆積,會使溶體破裂而吞噬掉自己的細胞,留下腦組織的空洞。接著這些PrP會繼續攻擊其他的細胞。而不正常的PrP也會在細胞外形成如阿茲海默症一樣的斑塊(plaques)。
2. PrP的基因沒有突變,但如有外來PrPsc,則PrPsc會誘發PrPc變成PrPsc。目前有一種結晶的理論,PrPSC進入神經細胞後,就取代原來的PrPC成為晶種,使得以後所製造出的蛋白質結構,會仿照PrPSC的形式,使細胞將充滿不正常的蛋白質。接著這些PrPsc就會如上一機制般的攻擊神經細胞,造成腦組織空洞。
當Prion進入腦神經後,在腦神經之間是經突觸由一個神經元傳至另一個神經。重要實驗發現
1. 1982年 Stanley Prusiner
(1)
將突變的PrP基因植入小鼠的染色體中,並使突變的PrP基因大量繁殖,結果基因轉殖的小鼠發病了,若將發病基因轉殖小鼠的腦萃取液打入另外一隻未發病基因轉殖的小鼠,則更快促使小鼠的發病及死亡
|
|
||
|
起先是健康的轉殖小鼠,再產生大量突變PrP之後病發死亡。 |
會製造少量病變PrP的轉殖小鼠在接受粹取液後,此轉殖小鼠也會發病。 |
另一隻會製造少量突變PrP的轉殖小鼠再接受粹取液後,也發病了。 |
==> PrP可以作為一種病原體
(2)
小鼠在發育的過程中顯然不需要PrPc,將小鼠的PrPc基因剔除(Knock-out mice, Prnp0/0),並將倉鼠的PrPsc 打入這些小鼠的腦中,這些小鼠並不會發病,也找不到大量的PrPsc,但是若將倉鼠的PrPsc打入含有倉鼠PrP基因的轉殖小鼠,PrPsc就像鑄模一樣,改變原來正常的prion結構,這感染的過程,發生在正常prion打開摺疊(unfolding)時,所以這些小鼠則會產生大量的PrPsc,並且發病。誘發的機制目前還不十分清楚,一些轉殖小鼠的實驗結果說明可能需要chaperone的幫忙
==>PrPc是使PrPsc增加的藍本,致病的PrP可以將正常的PrP誘發成致病性的PrP
==>異種間PrP不容易產生誘發,因為倉鼠和小鼠的PrP在254個胺基酸中有 16 個不同
若將CJD、GSS等病人的腦萃取液注入含有人類PrP基因的轉殖小鼠和含有人鼠混合PrP基因的轉殖小鼠,結果含有人鼠混合PrP基因的轉殖小鼠發病率較高
==>也許是人鼠混合PrPc比人類PrPc更容易被細胞內未知的chaperone認得
2. 2004年Stanley Prusiner
若使用基因轉植的技術利用大腸桿菌來表現prion蛋白(Recombinant mouse prion protein, recMoPrP),並將此蛋白質團塊植入小鼠腦中,則可以在小鼠腦中發現突變型的PrP堆積,而且這些突變型PrP還可以再用來感染其他小鼠
==>PrP僅需要突變型PrP就可以具有感染力
(但此實驗使用的基因轉殖的小鼠乃是特別容易患上相關病變的,因此並不能作為決定性的證據)
3. 2006年
(1)取野生型小鼠的骨隨細胞,並將其純化成含有prion及不含有prion的細胞,再將這些細胞分別植入被致命射線照射的小鼠體內,六個月後,在接受含有prion細胞的移植中,同時發現長生命期及短生命期的造血幹細胞,而在接受不含prion細胞的移植中,只發現短生命期的造血幹細胞
==>prion是野生型小鼠體中長生命期造血幹細胞的標記
(2)將野生型小鼠的骨隨細胞及不含有prion的小鼠骨髓細胞分別植入被致命射線照射的小鼠體內,當植入的幹細胞大量繁殖並且產生周邊血液細胞(peripheral blood cells)後,再將其植入另一隻被致命射線照射的小鼠體內,並重複這步驟三次。在每次的移植後,在接受不含prion骨髓細胞移植的小鼠的自我更生能力急遽下降,而由野生型小鼠細胞移植的小鼠的自我更生能力並不會下降。若使用可以產生prion的反轉錄病毒感染(retroviral infections)接受不含prion骨髓細胞移植的小鼠中,則小鼠的自我更生能力並不會下降
==>prion對造血幹細胞的自我更生有相當的必要性
對於prion如何影響造血幹細胞的自我更生能力,Harvey Lodish推測,prion可能是將細胞表面上的荷爾蒙結合並集中在一起,或是幫助細胞黏附到鄰近的細胞或細胞外基質上。這只要去看它是否黏附在其他的蛋白質或荷爾蒙上即可證明這個推測
Prion的治療方式
ð 抗體治療
有些抗體能夠在被培養的鼠腦細胞中清除這些普恩蛋白團塊。研究者表示它們似乎可以阻礙正常普恩蛋白轉變為異常型態的過程。在加州大學舊金山分校(UCSF) ,普恩蛋白研究先趨普西納 (Stanley Prusiner, 1997諾貝爾得主)的實驗室中領導計劃的 David Peretz表示:“我們研究的其中一株抗體可和正常普恩蛋白中,被認為會和病態普恩蛋白產生互動的區域結合。而其他的抗體則和普恩蛋白將會改變型狀的位置結合,以將之穩定。如果抗體能夠經過血液到達腦部,最終就可能將這些抗體發展成為疫苗,甚至是療法。”
ð 藥物治療
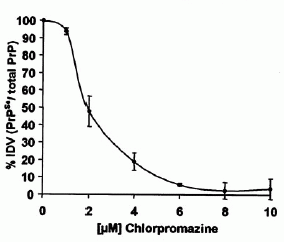
傳統藥物方面,目前研究的對象為奎那克林(quinacrine, 即 quinacrine hydrochlori,中文又稱虐滌平,治療瘧疾。)和氯普馬乙(chlorpromazinev,抗精神病藥物,有鎮靜作用,治療憂鬱症。),可阻礙惡性普恩蛋白團塊在受感染的陪有中的老鼠細胞中的形成。而且已知這兩種藥品可以穿過血腦屏障(Blood-Brain-Barrier),皆可讓PrPSc消失,又奎那克林比氯普馬乙效果好。
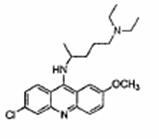
奎那克林 氯普馬乙

ð 基因治療
缺乏一個與免疫相關的基因DQ75的人感染變異型庫賈氏症(vCJD)的機會是一般人的三倍。倫敦的帝國醫學大學的John Collinge與同僚的發現在本研究中,五十名變異型庫賈式症患者(幾乎是已知病例的半數)中,只有12%有稱為“DQ7”的基因,而在正常人中約36%有此基因。要得到確實的結論,大約需要五百人的樣本,然而此數字遠大於目前已被記錄的病例總數。
ð 打散prion
目前研究人員表示,最具傳染性的是中長度的prion串列,較長或較短的似乎不是那麼大的問題。這個結果可說服專家重新思考如何治療變異庫賈氏症(vCJD)、阿茲海默症(Alzheimer's)及帕金森氏症(Parkinson's)等疾病。一些實驗性的治療致力於阻斷畸形蛋白質的形成及傳染,正使用於如變異庫賈氏症這類prion的疾病上。在動物實驗中,很多結果顯示,治療開始於症狀出現前似乎較有效。如果小心處理,那麼打散prion的叢集可能是另一個方法。問題是得將這些prion的叢集打散成較小的,而不殘留中間大小的。這些發現也包含了對阿茲海默症及帕金森氏症的治療。在這兩種疾病中,長的蛋白質在腦中成串形成。這些疾病的治療方法可能將分子鏈打斷成更有問題的中長度prion。
ð 以毒攻毒
美國UC Davis的兩位物理學家Cox和Singh用簡單的統計力學模型來模擬了普恩蛋白疾病的傳播。他們利用一組二維的晶格系統來進行模擬。他們發現一點點的prion可以在受到感染的神經元上製造出更多的prion。當到達足夠的數量之後便會分裂而讓其他的prion傳播出去到別的神經元上,最後造成宿主的死亡。事實上prion具有幾種不同的種類,例如有些對老鼠及倉鼠造成傷害但有些種類則是對倉鼠無害。這個模型發現,若是將無害的倉鼠普恩蛋白注入受感染的老鼠身上,可以使兩種不同的普恩蛋白彼此競爭,如此一來便可能可以減緩發病的時間。這種以毒攻毒的方法並不能保證能消滅這種疾病,但卻可以使類似CJD這種疾病不易在人身上發作。
資料來源:
大綱:
諾貝爾獎網站 http://nobelprize.org
輔仁大學理工學院生物技術研發中心http://brc.se.fju.edu.tw/nobelist/199x/p1997.htm
諾貝爾獎的故事 http://ortho.clmed.ncku.edu.tw/~cjlin/othernote/nobelstory.html
Prion的歷史:
J. Castilla et al., "In vitro generation of infectious scrapie prions," Cell, 121:195-206, April 22, 2005.
G. Legname et al., "Synthetic mammalian prions," Science, 305:673-6, July 30, 2004.
Prog. Biochem. Biophys.,”Prion疾病和”Protein only” 假說”, 生物化學與生物物理進展,95-105, February 31 2004
黃學謙,鍾作謙,”普利子疾病--庫賈氏病:一病例報告及文獻回顧”, 苗栗市大千綜合醫院內科神經科
科景科學新聞 http://www.sciscape.org
prion疾病病理:
國立成功大學圖書館醫學院分館 http://www.medlib.ncku.edu.tw/resources/giant.htm
行政院衛生署疾病管制局人畜共通傳染病資訊網
http://www.cdc.gov.tw/CDCzoo/Internet/decl/detail.aspx?uid=73&pid=5&AF_Id=33
高醫醫訊月刊第二十三卷第九期 http://www.kmuh.org.tw/www/kmcj/data/9302/14.htm
教育部顧問室『基礎科學前瞻性人才培育計畫』科普網站http://science.phy.ncu.edu.tw/
國家政策研究基金會--全球資訊網 http://www.npf.org.tw/
Sciscape 科景 http://www.sciscape.org/
(資料來源:memo.cgu.edu.tw/shu-er/95第2學期%20課堂報告/report/1b.doc)
(12月17日編輯完稿)
