{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
藥理學
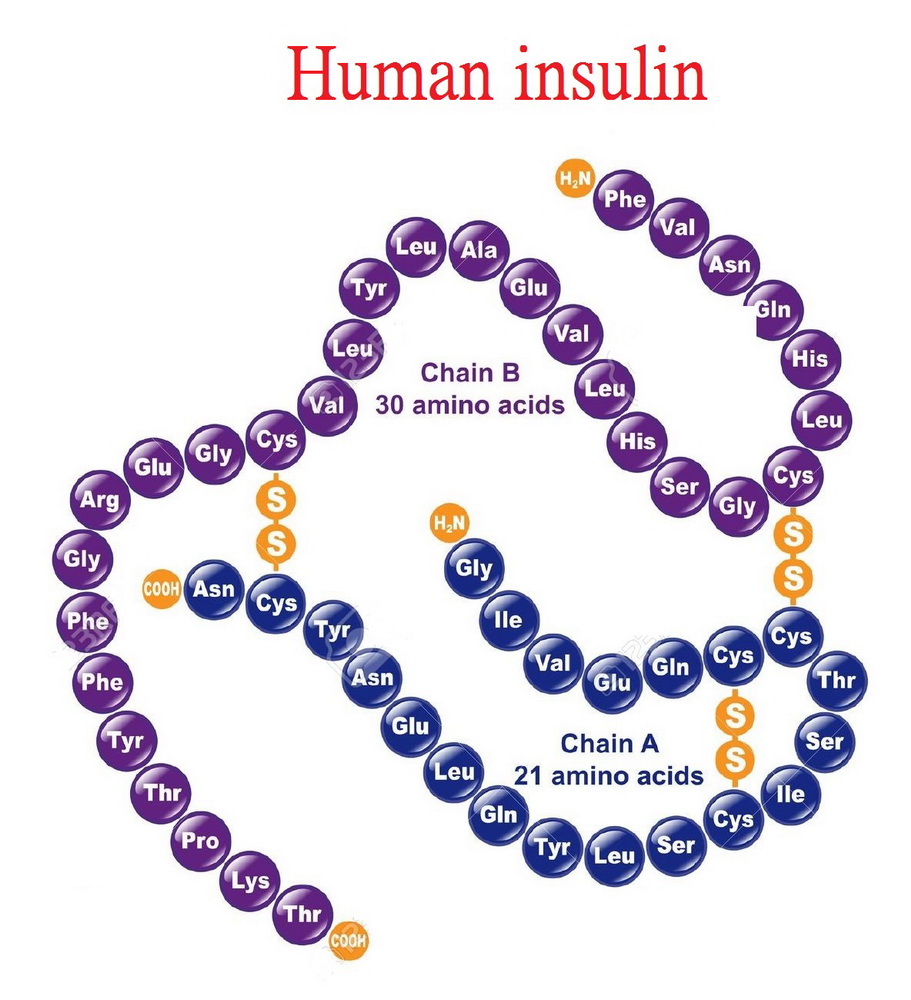
胰島素(insulin)是一個分子量為56kD的酸性蛋白質,
由兩條多肽鏈組成(A、B鏈),其間通過兩個二硫鏈以共價相聯。
藥用胰島素一般多由豬、牛胰腺提得。
目前可通過重組DNA技術利用大腸桿菌合成胰島素,
還可將豬胰島素β鏈第30位的丙氨酸用蘇氨酸代替而獲得人胰島素。
【體內過程】口服無效,因易被消化酶破壞,因此所有胰島素製劑都必須注射,皮下注射吸收快。
代謝快,t1/2為9∼10分鐘,但作用可維持數小時。
因其分布於組織後,與組織結合而在其中發揮作用。
主要在肝、腎滅活,經谷胱甘肽轉氨酶還原二硫鍵,再由蛋白水解酶水解成短肽或胺基酸,也可被腎胰島素酶直接水解。
嚴重肝腎功能不良者能影響其滅活。為延長胰島素的作用時間,可製成中效及長效製劑。
用鹼性蛋白質與之結合,使等電點提高到7.3,接近體液pH值,再加入微量鋅使之穩定,
這類製劑經皮下及肌內注射後,在注射部位發生沉澱,再緩慢釋放、吸收。
所有中、長效製劑均為混懸劑,不可靜注。見表36-1。
表36-1 胰島素製劑及其作用時間
分類
藥
物
注射途徑
作用時間(小時)
給藥時間
開始
高峰
維持
短效
正規胰島素(Regular Iusulin)
靜脈
立即 0.5
2
急救
皮下
0.5∼1
2∼3 6∼8
餐前0.5小時,3∼4次/日。
中效
低精蛋白鋅胰島素(Isophane Insulin)
皮下 2∼4 8∼12 18∼24
早餐或晚餐前1小時,一日∼2次
珠蛋白鋅胰島素(Globin Zinc Insulin)
皮下 2∼4 6∼10 12∼18
長效
精蛋白鋅胰島素(Protamine Zinc Insulin)
皮下 3∼6 16∼18 24∼36
早餐或晚餐前1小時,一日1次
【藥理作用】胰島素對代謝過程具有廣泛的影響。
1.糖代謝 胰島素可增加葡萄糖的轉運,加速葡萄糖的氧化和酵解,促進糖原的合成和貯存,抑製糖原分解和異生而降低血糖。
2.脂肪代謝 胰島素能增加脂肪酸的轉運,促進脂肪合成並抑制其分解,減少游離脂肪酸和酮體的生成。
3.蛋白質代謝 胰島素可增加胺基酸的轉運和蛋白質的合成(包括mRNA的轉錄及翻譯),同時又抑制蛋白質的分解。
【作用機制】已知胰島素受體為一糖蛋白,是由兩個13kD的α-亞單位及兩個90kD的β-亞單位組成的大分子蛋白複合物。
α-亞單位在胞外,含胰島素結合部位,β-亞單位為跨膜蛋白,其胞內部分含酪氨酸蛋白激酶,
胰島素需與靶細胞膜受體結合後,才能產生一系列的生物效應,對產生效應的機制有以下假說,
一認為胰島素可誘導第二信使的形成,它們模擬或具有胰島素樣的活性。
二認為胰島素與α-亞單位結合,移入胞內後可激活酪氨酸蛋白激酶,繼而催化受體蛋白自身及胞內其他蛋白的酪氨酸殘基磷酸化,
因而啟動了磷酸化的連鎖反應(phosphorylation
cascade)。
三認為胰島素可使葡萄糖載體蛋白(glucose transporter)和其他蛋白質從胞內重新分布到胞膜,從而加速葡萄糖的轉運。
胰島素受體的基本結構
圖36-1
胰島素受體的基本結構 圖註:α、β=亞單位 -S-S-=雙硫鍵 =糖基化部位
胰島素與α-亞單位相結合;β-亞單位胞內部分含酪氨酸蛋白激酶
【臨床應用】胰島素仍是治療胰島素依賴型糖尿病(insulin-dependent diabetis mellitus,IDDM)的唯一藥物,
對胰島素缺乏的各型糖尿病均有效。
主要用於下列情況:
①重症糖尿病(IDDM,I型);
②非胰島素依賴型糖尿病(noninsulin dependent diabetis mellitus,NIDDM)經飲食控制或用口服降血糖藥未能控制者;
③糖尿病發生各種急性或嚴重併發症者,如酮症酸中毒及非酮症高血糖高滲性昏迷(要建立和維持電解質的平衡)。
④合併重度感染、消耗性疾病、高熱、妊娠、創傷以及手術的各型糖尿病。
【不良反應】
1.過敏反應
多數為使用牛胰島素所致,它作為異體蛋白進入人體後可產生相應抗體如IgE並引起過敏反應。
一般反應輕微而短暫,偶可引起過敏休克。可用豬胰島素代替,因其與人胰島素較為接近。
2.低血糖症
為胰島素過量所致,正規胰島素能迅速降低血糖,出現飢餓感、出汗、心跳加快、焦慮、震顫等症状,嚴重者引起昏迷、
驚厥及休克,甚至腦損傷及死亡。長效胰島素降血糖作用較慢,不出現上述症状,而以頭痛和精神情緒、運動障礙為主要表現。
為防止低血糖症的嚴重後果,應教會病人熟知反應,以便及早發現和攝食,或飲用糖水等。
嚴重者應立即靜脈注射50%葡萄糖。必須在糖尿病患者中鑒別低血糖昏迷和酮症酸中毒性昏迷及非酮症性糖尿病昏迷。
3.胰島素耐受性
產生急性耐受常由於並發感染、創傷、手術、情緒激動等應激狀態所致。
此時血中抗胰島素物質增多,或因酮症酸中毒時,血中大量游離脂肪酸和酮體的存在妨礙了葡萄糖的攝取和利用。
出現急性耐受時,需短時間內增加胰島素劑量達數千單位。
產生慢性耐受的原因較為複雜(系指每日需用200U以上的胰島素並且無併發症者)。
可能是體內產生了抗胰島素受體抗體(AIRA),對此可用免疫抑制劑控制症状,能使患者對胰島素的敏感性恢復正常;
也可能是胰島素受體數量的變化,如高胰島素血症時,靶細胞膜上胰島素受體數目減少;還可能是靶細胞膜上葡萄糖轉運系統失常。
此時換用其他動物胰島素或改用高純度胰島素,並適當調整劑量常可有效。
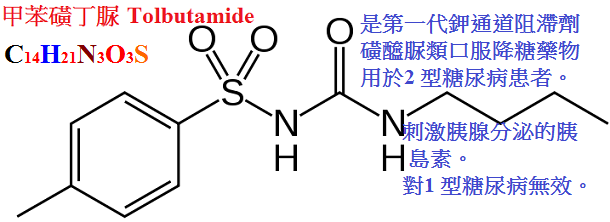
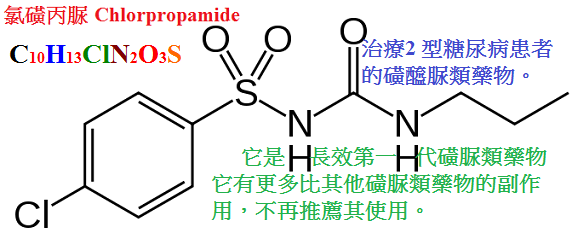
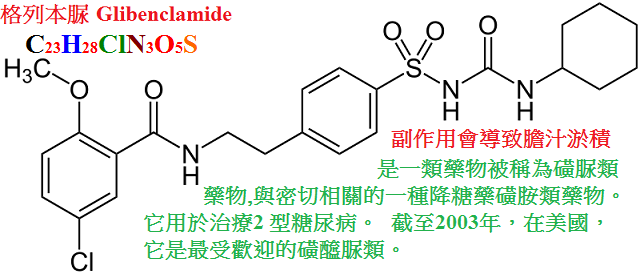
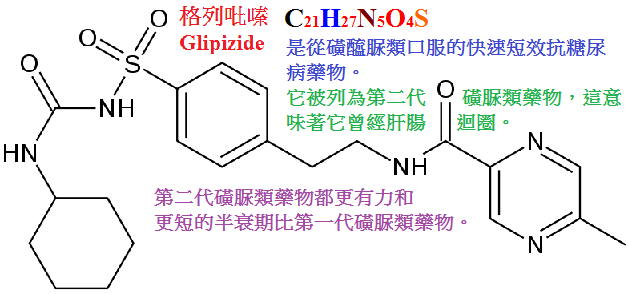
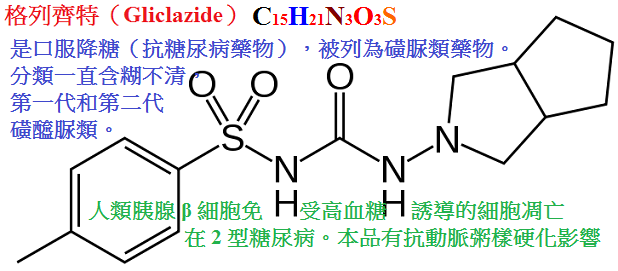
甲苯磺丁脲(tolbutamid,D860,甲糖寧)、氯磺丙脲(chlorpropamide),格列本脲(glyburide,glibenclamide,優降糖),
格列吡嗪(glipizide,吡磺環已脲),格列齊特(gliclazipe,達美康)等,化學結構如下:
表36-2 磺醯脲類藥物的化學結構
【藥理作用及作用機制】胰島β細胞膜含有磺醯脲受體及與之相偶聯的ATP敏感的鉀通道[Ik(ATP)],以及電壓依賴性的鈣通道。
當磺醯脲類藥物與其受體相結合後,可阻滯Ik(ATP)而阻鉀外流,致使細胞膜去極化,增強電壓依賴性鈣通道開放,胞外鈣內流。
胞內游離鈣濃度增加後,觸發胞吐作用及胰島素的釋放。
長期服用且胰島素已恢復至給藥前水平的情況下,其降血糖作用仍然存在,這可能與抑制胰高血糖素的分泌,
提高靶細胞對胰島素的敏感性有關。也可能與增加靶細胞膜上胰島素受體的數目和親和力有關。
【體內過程】磺醯脲類藥物在胃腸道吸收迅速而完全,與血漿蛋白結合率很高。
其中多數藥物在肝內氧化成羥基化合物,並迅速從尿中排出。磺醯脲類藥物的藥代動力學見表36-3。
甲苯磺丁脲作用最弱、維持時間最短,而氯磺丙脲t1/2最長,且排泄慢、每日只需給藥一次。
新型磺醯脲類作用較強,可維持24小時,每日只需給藥1∼2次。
表36-3 磺醯脲類藥物的藥代動力學參數
藥
物
給藥
途徑
效強
血漿蛋白
結合
作用持續時間(h) t1/2
代謝途徑
排泄(經肝、腎)
甲苯磺丁脲
口服 +
>90% 4∼6
3∼5
氧化 95%
氯磺丙脲
口服 +++
>90% 60 24∼48
不代謝
90%
格列本脲
口服 ++++
>90% 24 10∼16
氧化 50%
格列吡嗪
口服 ++++
>90% 24 3∼7
氧化 90%
【臨床應用】
1.糖尿病 用於胰島功能尚存的非胰島素依賴型糖尿病且單用飲食控制無效者。
對胰島素產生耐受的患者用後可刺激內源性胰島素的分泌而減少胰島素的用量。
2.氯磺丙脲能促進抗利尿素的分泌,可治療尿崩症。
【不良反應】常見不良反應為胃腸不適、噁心、腹痛、腹瀉。
大劑量氯磺丙脲還可引起中樞神經系統症状,如精神錯亂、嗜睡、眩暈、共濟失調。
也可引起粒細胞減少和膽汁鬱積性黃疸及肝損害,一般在服藥後1∼2個月內發生。
因此需定期檢查肝功能和血象。
較嚴重的不良反應為持久性的低血糖症,常因藥物過量所致,尤以氯磺丙脲為甚。
老人及肝、腎功能不良者較易發生,故老年糖尿病人不宜用氯磺丙脲。新型磺醯脲類較少引起低血糖。
【藥物相互作用】由於磺醯脲類有較高的血漿蛋白結合率,因此在蛋白結合上能與其他藥物
(如保泰松、水楊酸鈉、吲哚美辛、青黴素、雙香豆素等)發生競爭,使游離藥物濃度上升而引起低血糖反應。
此外,氯丙嗪、糖皮質激素、噻嗪類利尿藥、口服避孕藥均可降低磺醯脲類藥物的降血糖作用。
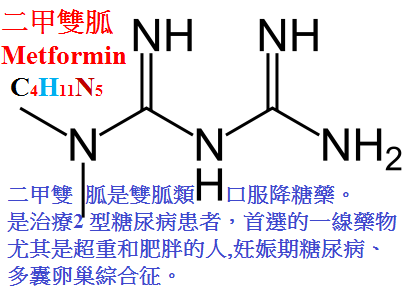
國內應用的有甲福明(metformin,二甲雙胍)、苯乙福明(phenformine,苯乙雙胍)。
甲福明作用短,在體內不與蛋白結合,不被代謝,從尿中排出。
其作用機制可能是降低食物吸收及糖原異生、促進組織攝取葡萄糖等。
主要用於輕症糖尿病患者,尤適用於肥胖者,單用飲食控制無效者。
不良反應為食慾下降、噁心、腹部不適、腹瀉等,危及生命的不良反應為乳酸血症,尤以苯乙福明的發生率高。
與苯乙福明相比,甲福明一般不引起乳酸血症,應用較廣。
α-葡萄糖甙酶抑制劑是一類新型口服降血糖藥,
其中阿卡波糖(acarbose)已用於臨床,
其降血糖的機制是:
在小腸上皮刷狀緣與碳水化合物競爭水解碳水化合物的酶,
從而減慢水解及產生葡萄糖的速度並延緩葡萄糖的吸收。
血糖峰值降低。主要副作用為胃腸道反應。
服藥期間應增加碳水化合物的比例,
並限制單糖的攝入量,以提高藥物的療效。
胰島素(insulin,正規胰島素,regular insulin)
低精蛋白鋅胰島素(isophane
insulin)
珠蛋白鋅胰島素(globin zinc insulin)
精蛋白鋅胰島素(protamine zinc
insulin)
甲苯磺丁脲(tolbutamide,D860,甲糖寧)
氯磺丙脲(chlorpropamide,P-607)
格列本脲(glibenclamide,HB-419)
甲福明(metformin,Diabex,DMBG,二甲雙胍,降糖片)
抗菌藥物對病原菌具有抑制或殺滅作用,是防治細菌感染性疾病的一類藥物。
細菌和其他微生物、寄生蟲及癌細胞所致疾病的藥物治療統稱為化學治療學(chemotherapy,簡稱化療)。
化學治療學的目的是研究、應用對病原體有選擇毒性(即強大殺滅作用),
而對宿主無害或少害的藥物以防治病原體所引起的疾病。
機體、抗菌藥物及病原微生物的相互作用關係
圖37-1
機體、抗菌藥物及病原微生物的相互作用關係
在應用化療藥物治療感染性疾病過程中,應注意機體、病原體與藥物三者的相互關係(圖37-1)。
感染性疾病的罹患與康復是微生物與機體相互鬥爭的過程。病原微生物在疾病的發生上無疑起著重要作用。
但病原體不能決定疾病的全過程,人體的反應性、免疫狀態和防禦功能對疾病的發生、發展與轉歸也有重要作用。
當機體防禦功能佔主導地位時,就能戰勝致病微生物,使它不能致病,或發病後迅速康復。
抗菌藥物的抑菌或殺菌作用是制止疾病發展與促進康復的外來因素,
為機體徹底消滅病原體和導致疾病痊癒創造有利條件。
事物總是有兩面性的,矛盾是不斷轉化的。在某種條件下微生物可產生耐藥性,而使藥物失去抗菌效果;
在治療中藥物的治療作用是主要的,但使用不當時,藥物可產生不良反應,影響患者健康,甚至使治療失敗。
常用術語
抗菌譜 每種抗菌藥物都有一定的抗菌範圍,稱為抗菌譜。
某些抗菌藥物僅作用於單一菌種或局限於一屬細菌,其抗菌譜窄,如異煙肼只對抗酸分支桿菌有效。
另一些藥物抗菌範圍廣泛稱之為廣譜抗菌藥,如四環素和氯黴素,它們不僅對革蘭陽性細菌和革蘭陰性細菌有抗菌作用,
且對衣原體、肺炎支原體、立克次體及某些原蟲等也有抑制作用。近年新發展的青黴素類和頭孢菌素類抗生素也有廣譜抗菌作用。
抗菌活性
抗菌活性是指藥物抑制或殺滅微生物的能力。一般可用體外與體內(化學實驗治療)兩種方法來測定。
體外抗菌試驗對臨床用藥具有重要意義。能夠抑制培養基內細菌生長的最低濃度稱之為最低抑菌濃度(MIC);
能夠殺滅培養基內細菌的最低濃度稱之為最低殺菌濃度(MBC)。
抑菌藥 是指僅有抑制微生物生長繁殖而無殺滅作用的藥物,如四環素等。
殺菌藥 這類藥不僅能抑制微生物生長繁殖,而且能殺滅之,如青黴素類、氨基甙類等。
化療指數
理想的化療藥物一般必須具有對宿主體內病原微生物有高度選擇性的毒性,而對宿主無毒性或毒性很低,
最好還能促進機體防禦功能並能與其他抗菌藥物聯合應用消滅病原體。
化療藥物的價值一般以動物半數致死量(LD50)和治療感染動物的半數有效量(ED50)之比,
或5%致死量(LD5)與95%有效量(ED95)的比來衡量。這一比例關係稱為化療指數。
化療指數愈大,表明藥物的毒性愈小,療效愈大,臨床應用的價值也可能愈高
但化療指數高者並不是絕對安全,如幾無毒性的青黴素仍有引起過敏休克的可能。
抗菌藥物的作用機制,現多以干擾細菌的生化代謝過程來解釋。茲將幾種主要方式(圖37-2)簡介如下:
細菌結構與抗菌藥作用部位示意圖
圖37-2
細菌結構與抗菌藥作用部位示意圖
目錄
1
一、抗葉酸代謝
2
二、抑制細菌細胞壁合成
3
三、影響胞漿膜的通透性
4
四、抑制蛋白質合成
一、抗葉酸代謝
磺胺類與甲氧苄啶(TMP)可分別抑制二氫葉酸合成酶與二氫葉酸還原酶,
妨礙葉酸代謝,最終影響核酸合成,從而抑制細菌的生長和繁殖(見42章)。
二、抑制細菌細胞壁合成
細菌細胞膜外是一層堅韌的細胞壁,能抗禦菌體內強大的滲透壓,具有保護和維持細菌正常形態的功能。
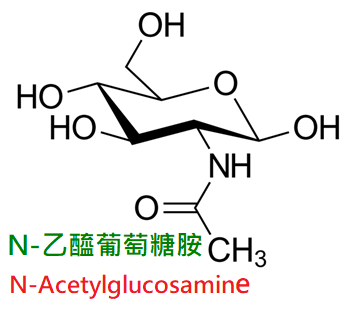
細菌細胞壁主要結構成分是胞壁粘肽,由N-乙醯葡萄糖胺(GNAc)和與五肽相連的N-乙醯胞壁酸(MNAc)重複交替聯結而成。
胞壁粘肽的生物合成可分為胞漿內、胞漿膜與胞漿外三個階段。
胞漿內粘肽前體的形成可被磷黴素與環絲氨酸所阻礙。
磷黴素抑制有關酶系阻礙N-乙醯胞壁酸的形成;環絲氨酸通過抑制D-丙氨酸的消旋酶和合成酶阻礙了N-乙醯胞壁酸五肽的形成。
胞漿膜階段的粘肽合成可被萬古黴素和桿菌肽所破壞,
它們能分別抑制MNAc-五肽與脂載體結合併形成直鏈十肽二糖聚合物和聚合物轉運至膜外受體的過程及脫磷酸反應。
青黴素與頭孢菌素類抗生素則能阻礙直鏈十肽二糖聚合物在胞漿外的交叉聯接過程。
青黴素等的作用靶位是胞漿膜上的青黴素結合蛋白(PBPs),表現為抑制轉肽酶的轉肽作用,從而阻礙了交叉聯接。
能阻礙細胞壁合成的抗生素可導致細菌細胞壁缺損。由於菌體內的高滲透壓,在等滲環境中水分不斷滲入。
致使細菌膨脹、變形,在自溶酶影響下,細菌破裂溶解而死亡。
三、影響胞漿膜的通透性
細菌胞漿膜主要是由類脂質和蛋白質分子構成的一種半透膜,具有滲透屏障和運輸物質的功能。
多粘菌素類抗生素具有表面活性物質,能選擇性地與細菌胞漿膜中的磷酯結合;
而制黴菌素和二性黴素等多烯類抗生素則僅能與真菌胞漿膜中固醇類物質結合。
它們均能使胞漿膜通透性增加,導致菌體內的蛋白質、核苷酸、胺基酸、糖和鹽類等外漏,從而使細菌死亡。
四、抑制蛋白質合成
細菌為原核細胞,其核蛋白體為70S,由30S和50S亞基組成,哺乳動物是真核細胞,其核蛋白體為80S,由40S與60S亞基構成,
因而它們的生理、生化與功能不同,抗菌藥物對細菌的核蛋白體有高度的選擇性毒性,
而不影響哺乳動物的核蛋白體和蛋白質合成。多種抗生素能抑制細菌的蛋白質合成,但它們的作用點有所不同。
①能與細菌核蛋白體50S亞基結合,使蛋白質合成呈可逆性抑制的有氯黴素、林可黴素和大環內酯類抗生素(紅霉素等)。
②能與核蛋白體30S亞基結合而抑菌的抗生素如四環素能阻止氨基醯tRNA向30S亞基的A位結合,從而抑制蛋白質合成。
③能與30S亞基結合的殺菌藥有氨基甙類抗生素(鏈黴素等)。
它們的作用是多環節的。影響蛋白質合成的全過程,因而具有殺菌作用。
五、抑制核酸代謝
喹諾酮類藥物能抑制DNA的合成,利福平能抑制以DNA為模板的RNA多聚酶。
細菌的耐藥性又稱抗藥性,一般是指細菌與藥物多次接觸後,對藥物的敏感性下降甚至消失,致使藥物對耐藥菌的療效降低或無效。
一、耐藥性產生機制
1.產生滅活酶
滅活酶有兩種,一是水解酶,如β-內醯胺酶可水解青黴素或頭孢菌素。
該酶可由染色體或質體介導,某些酶的產生為體質性(組構酶);某些則可經誘導產生(誘導酶)。
二是鈍化酶又稱合成酶,可催化某些基團結合到抗生素的OH基或NH2基上,使抗生素失活。
多數對氨基甙類抗生素耐藥的革蘭陰性桿菌能產生質體介導的鈍化酶,
如乙醯轉移酶作用於NH2基上,磷酸轉移酶及核苷轉移酶作用於OH基上。
上述酶位於胞漿膜外間隙,氨基甙類被上述酶鈍化後不易與細菌體內的核蛋白體結合,從而引起耐藥性。
2.胞漿膜通透性
細菌可通過各種途徑使抗菌藥物不易進入菌體,如革蘭陰性桿菌的細胞外膜對青黴素G等有天然屏障作用;
綠膿桿菌和其他革蘭陰性桿菌細胞壁水孔或外膜非特異性通道功能改變引起細菌對一些廣譜青黴素類、
頭孢菌素類包括某些第三代頭孢菌素的耐藥;
細菌對四環素耐藥主要由於所帶的耐藥質體可誘導產生三種新的蛋白,阻塞了細胞壁水孔,使藥物無法進入。
革蘭陰性桿菌對氨基甙類耐藥除前述產生鈍化酶外,也可由於細胞壁水孔改變,使藥物不易滲透至細菌體內。
3.細菌體內靶位結構的改變
鏈黴素耐藥株的細菌核蛋白體30S亞基上鏈黴素作用靶位P10蛋白質發生改變;
利福平的耐藥性是細菌RNA多聚酶的β'亞基發生改變,使其與藥物的結合力降低而耐藥。
由質體介導的對林可黴素和紅霉素的耐藥性,系細菌核蛋白體23S亞基的腺嘌呤甲基化,使藥物不能與細菌結合所致。
某些肺炎球菌、淋球菌對青黴素G耐藥,以及金葡菌對甲氧苯青黴素耐藥,
乃因經突變引起青黴素結合蛋白(PBPs)改變,使藥物不易與之結合。
這種耐藥株往往對其他青黴素(如苯唑或鄰氯青黴素)和頭孢菌素類也都耐藥。
4.其他
細菌對磺胺類的耐藥,可由對藥物具拮抗作用的底物PABA的產生增多所致;也可能通過改變對代謝物的需要等途徑。
二、避免細菌耐藥性的措施
為了克服細菌對藥物產生耐藥性,
臨床醫生要注意抗菌藥物的合理應用,給予足夠的劑量與療程,必要的聯合用藥和有計劃的輪換供藥。
此外,醫藥學專家還應努力開發新的抗菌藥物,改造化學結構,使其具有耐酶特性或易於透入菌體。
由於抗菌藥的使用,過去許多致死性的疾病已得到控制。
但隨著抗菌藥物的廣泛使用,特別是濫用,也給治療帶來許多新問題,
如毒性反應、過敏反應、二重感染、細菌產生耐藥性等。
因此,合理使用抗菌藥物日益受到重視。
一、抗菌藥臨床應用的基本原則
(一)嚴格按照適應證選藥
每一種抗菌藥物各有不同抗菌譜與適應證。臨床診斷、細菌學診斷和體外藥敏試驗可作為選藥的重要參考。
表37-1供選藥時參考。
此外,還應根據病人全身情況,肝、腎功能,感染部位,藥物代謝動力學特點,
細菌產生耐藥性的可能性、不良反應和價格等方面因素綜合考慮。
表37-1 藥敏試驗中的抗菌藥物選擇
腸桿菌科
假單胞菌屬
金葡菌
腸球菌屬
流感桿菌
第一線 氨苄西林 哌拉西林 頭孢噻吩 青黴素 氨苄西林-舒巴坦 羧苄西林 慶大黴素 紅霉素
頭孢呋新
第二線
頭孢呋新
諾氟沙星
慶大黴素
萬古黴素
氯黴素
復方SMZ-TMP
阿米卡星 哌拉西林
頭孢他啶
(或頭孢哌酮)
利福平
第三代頭孢菌素 復方SMZ-TMP
諾氟沙星
呋喃妥因
呋喃妥因
紅霉素
(二)病毒性感染和發熱原因不明者
感冒、上呼吸道感染等病毒性疾病,發病原因不明者(除病情嚴重並懷疑為細菌感染外)不宜用抗菌藥,
否則可使臨床症状不典型和病原菌不易被檢出,以致延誤正確診斷與治療。
(三)抗菌藥劑量
劑量要適當,療程應足夠。劑量過小,不但無治療作用,反易使細菌產生耐藥性;
劑量過大,不僅造成浪費,還會帶來嚴重的毒副作用。療程過短易使疾病複發或轉為慢性。
(四)皮膚粘膜等局部感染
應盡量避免局部應用抗菌藥,因其易發生過敏反應和耐藥菌的產生。
(五)預防應用及聯合應用
對此均應嚴格掌握適應證,抗菌藥物的預防應用僅限於少數情況,如經臨床實踐證明確有效果者;
聯合用藥,也必須謹慎掌握指征、權衡利弊。
二、抗菌藥的聯合應用
(一)抗菌藥聯合應用的意義
聯合用藥的主要優點是:
①發揮藥物的協同抗菌作用以提高療效;
②延遲或減少耐藥菌的出現;
③對混合感染或不能作細菌學診斷的病例,聯合用藥可擴大抗菌範圍;
④聯合用藥可減少個別藥劑量,從而減少毒副反應。
濫用抗菌藥物的聯合應用,可能產生不利後果:如增加不良反應發生率;容易出現二重感染;
耐藥菌株更加增多;浪費藥物;給人一種虛偽的安全感染,延誤正確治療。
(二)聯合用藥的指征
聯合用藥的指征有:
①病原菌未明的嚴重感染;
②單一抗菌藥物不能控制的嚴重混合感染,如腸穿孔後腹膜炎的致病菌常有多種需氧菌和厭氧菌等;
③單一抗菌藥物不能有效控制的感染性心內膜炎或敗血症;
④長期用藥細菌有可能產生耐藥者,如結核、慢性尿路感染、慢性骨髓炎等;
⑤用以減少藥物毒性反應,如兩性黴素B和氟胞嘧啶合用治療深部真菌,前者用量可減少,從而減少毒性反應;
⑥臨床感染一般用二藥聯用即可,常不必要三藥聯用或四藥聯用。
(三)聯合用藥可能產生結果
兩種抗菌藥聯合應用在體外或動物實驗中可獲得無關、相加、協同(增強)和拮抗等四種效果。
抗菌藥物依其作用性質可分為四大類:
一類為繁殖期殺菌,如青黴素類、頭孢菌素類等;
二類為靜止期殺菌,如氨基甙類、多粘菌素等,它們對靜止期、繁殖期細菌均有殺滅作用;
三類為速效抑菌,如四環素類、氯黴素類與大環內酯類抗生素等、
四類為慢效抑菌劑,如磺胺類等。
第一類和第二類合用常可獲得協同(增強)作用,例如青黴素與鏈黴素或慶大黴素合用治療腸球菌心內膜炎;
青黴素破壞細菌細胞壁的完整性,有利於氨基甙類抗生素進入細胞內發揮作用。
第一類與第三類合用可能出現拮抗作用。例如青黴素類與氯黴素或四環素類合用。
由於後二藥使蛋白質合成迅速被抑制,細菌處於靜止狀態,致使繁殖期殺菌的青黴素干擾細胞壁合成的作用不能充分發揮,使其抗菌活性減弱。
第二類和第三類合用可獲得增強或相加作用。
第四類慢效抑菌藥與第一類可以合用,例如,治療流行性腦膜炎時,青黴素可以和磺胺嘧啶合用而提高療效。
應該指出上述資料多來自體外與動物試驗在特定條件下的觀察,與臨床實際不盡相同,僅供參考。
聯合用藥產生的作用也可因不同菌種和菌株而異,藥物劑量和給藥順序也會影響效果。
三、肝腎功能損害時抗菌藥的應用
(一)腎功能損害
腎功能減退時,應用主要經腎排泄的藥物宜減量或延長給藥時間。
對腎有毒的藥物,如兩性黴素B、萬古黴素及氨基甙類等,宜避免使用。
對腎功能無損害或損害不大的藥物在一般情況下,可按常規給藥,但要求肝功能必須正常。
腎功能輕、中和重度減退的給藥量分別為正常劑量的2/3∼1/2,1/2∼1/5和1/5∼1/10。
(二)肝功能障礙的影響
肝功能減退者,應避免使用或慎用氯黴素、林可黴素、紅霉素、利福平、四環素類等。
早產和新生兒的肝臟對氯黴素的解毒功能較低,氯黴素列為禁用。
β-內醯胺類抗生素(β-lactams)系指化學結構中具有β-內醯胺環的一大類抗生素,
包括臨床最常用的青黴素與頭孢菌素,以及新發展的頭黴素類、硫黴素類、單環β-內醯胺類等其他非典型β-內醯胺類抗生素。
此類抗生素具有殺菌活性強、毒性低、適應症廣及臨床療效好的優點。
本類藥化學結構,特別是側鏈的改變形成了許多不同抗菌譜和抗菌作用以及各種臨床藥理學特性的抗生素。
(一)抗菌作用機制
各種β-內醯胺類抗生素的作用機制均相似,都能抑制胞壁粘肽合成酶,
即青黴素結合蛋白(penicillin binding proteins,PBPs),
從而阻礙細胞壁粘肽合成,使細菌胞壁缺損,菌體膨脹裂解(胞壁粘肽合成過程見三十七章)。
除此之外,對細菌的致死效應還應包括觸發細菌的自溶酶活性,缺乏自溶酶的突變株則表現出耐藥性。
哺乳動物無細胞壁,不受β-內醯胺類藥物的影響,因而本類藥具有對細菌的選擇性殺菌作用,對宿主毒性小。
近十多年來已證實細菌胞漿膜上特殊蛋白PBPs是β-內醯胺類藥的作用靶位,PBPs的功能及與抗生素結合情況歸納於圖38-1。
各種細菌細胞膜上的PBPs數目、分子量、對β-內醯胺類抗生素的敏感性不同,但分類學上相近的細菌,其PBPs類型及生理功能則相似。
例如大腸桿菌有7種PBPs,PBP1A,PBP1B與細菌延長有關,青黴素、氨苄西林、頭孢噻吩等與PBP1A、PBP1B有高度親和力,
可使細菌生長繁殖和延伸受抑制,並溶解死亡,PBP2與細管形狀有關,美西林、棒酸與硫黴素(亞胺培南)能選擇性地與其結合,
使細菌形成大圓形細胞,對滲透壓穩定,可繼續生幾代後才溶解死亡。
PBP3功能與PBP1A相同,但量少,與中隔形成,細菌分裂有關,多數青黴素類或頭孢菌素類抗生素主要與PBP1和(或)PBP3結合,
形成絲狀體和球形體,使細菌發生變形萎縮,逐漸溶解死亡。
PBP1,2,3是細菌存活、生長繁殖所必需,PBP4,5,6;
與羧肽酶活性有關,對細菌生存繁殖無重要性,抗生素與之結合後,對細菌無影響。
大腸桿菌PBPs的酶功能及與其結合的抗生素的作用
圖38-1 大腸桿菌PBPs的酶功能及與其結合的抗生素的作用
(二)影響β-內醯胺類抗菌作用素
革蘭陽性菌與陰性菌的結構差異甚大,β-內醯胺類各藥與母核相聯接的側鏈不同可影響其親脂性或親水性。
有效藥物必需能進入菌體作用於細胞膜上的靶位PBPs。影響抗菌作用的主要因素:
①藥物透過革蘭陽性菌細胞壁或陰性菌脂蛋白外膜(即第一道穿透屏障)的難易;
②對β-內醯胺酶(第二道酶水解屏障)的穩定性;
③對抗菌作用靶位PBPs的親和性。
根據這些因素,目前臨床應用的β-內醯胺類對革蘭陽性與陰性菌的作用大致有6種類型(見圖38-2)。
革蘭陽性與陰性菌的結構及β內醯胺類藥的穿透情況及其對β-內醯胺酶與胞壁合成酶(PBPs)的關係示意圖
圖38-2 革蘭陽性與陰性菌的結構及β內醯胺類藥的穿透情況及其對β-內醯胺酶與胞壁合成酶(PBPs)的關係示意圖
Ⅰ類為青黴素及口服青黴素V易透過革蘭陽性菌胞壁粘肽層,
但它們不能透過革蘭陰性菌糖蛋白磷脂外膜,因而屬窄譜的僅對革蘭陽性菌有效。
Ⅱ類包括有氨苄西林、羧苄西林、醯脲類青黴素、亞胺培南及若干頭孢菌素,能適度透過革蘭陽性菌的胞壁粘肽層,
對革蘭陰性菌的外膜透過性則很好,因而是廣譜抗菌藥物。
Ⅲ類為青黴素等容易被革蘭陽性菌的胞外β-內醯胺酶即青黴素酶破壞滅活的青黴素類,對產酶菌往往表現明顯的耐藥性。
Ⅳ類為異噁唑類青黴素、頭孢菌素一、二代及亞胺培南等對青黴素酶穩定,對革蘭陽性的產酶菌有效,
但對染色體突變而改變的PBPs結構,可使藥物與PBPs的親和力下降或消失,因而無效。
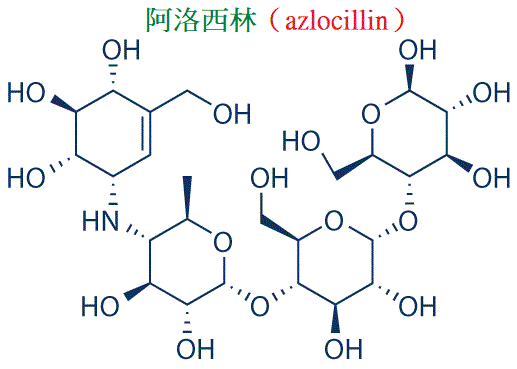
Ⅴ類包括醯脲類青黴素(阿洛西林與美洛西林等)、羧苄青黴素及頭孢菌素一、二代,
當胞膜外間隙的β-內醯胺酶少量存在時有抗菌效果,大量酶存在時,則被破壞而無效。
Ⅵ類包括第三代頭孢菌素、氨曲南、亞胺培南等對β-內醯胺酶十分穩定,即使大量β-內醯胺酶存在時仍然有效,
但對因染色體突變而改變了的PBPs則無效,加用氨基甙類抗生素也仍然無效。
(三)細菌耐藥機制
細菌對β-內醯胺類抗生素耐藥機制可概括為:
①細菌產生β-內醯胺酶(青黴素酶、頭孢菌素酶等)使易感抗生素水解而滅活;
②對革蘭陰性菌產生的β-內醯胺酶穩定的廣譜青黴素和第二、三代頭孢菌素,
其耐藥發生機制不是由於抗生素被β-內醯胺酶水解,
而是由於抗生素與大量的β-內醯胺酶迅速、牢固結合,使其停留於胞膜外間隙中,因而不能進入靶位(PBPs)發生抗菌作用。
此種β-內醯胺酶的非水解機制又稱為「牽制機制」(trappingmechanism);
③PBPs靶蛋白與抗生素親和力降低、PBPs增多或產生新的PBPs均可使抗生素失去抗菌作用。
例如MRSA(methicillin
resistant Staphylococcusaureus)具有多重耐藥性,其產生機制是PBPs改變的結果,
高度耐藥性系由於原有的PBP2與PBP3之間產生一種新的PBP2'(即PBP2a),
低、中度耐藥系由於PBPs的產量增多或與甲氧西林等的親和力下降所致;
④細菌的細胞壁或外膜的通透性改變,使抗生素不能或很少進入細菌體內到達作用靶位。
革蘭陰性菌的外膜是限制β-內醯胺類抗生素透入菌體的第一道屏障。
近年研究已證實抗生素透入外膜有非特異性通道與特異性通道兩種。
大腸桿菌K-12外膜有親水性的非特異性孔道蛋白(porin)為三聚體結構,
有二個孔道蛋白,即OmpF與OmpC,其合成由OmpB3基因調控。
OmpF的直徑為1nm,許多重要的β-內醯胺類抗生素大多經過此通道擴散入菌體內。
鼠傷寒桿菌OmpF與OmpC缺陷突變株對頭孢噻啶的通透性要比野生株小10倍,因而耐藥。
僅含微量OmpF與OmpC的大腸桿菌突變株,對頭孢唑啉、頭孢噻吩的透入也較野生株成倍降低,
其MIC明顯增高,也出現耐藥。
綠膿桿菌對β-內醯胺類抗生素耐藥性的產生已證明是由於外膜非特異性孔道蛋白OprF缺陷而引起的。
革蘭陰性外膜的特異性通道,在綠膿桿菌耐亞胺培南的突變株已證明系由於外膜缺失一種分子量為45∼46kD蛋白OprD。
如將此OprD重組於缺陷OprD的突變株外膜蛋白脂質體中,
又可使亞胺培南透過性增加5倍以上,其MIC也相應地降低,於是細菌的耐藥性消除。
⑤由於細菌缺少自溶酶而出現細菌對抗生素的耐藥性,即抗生素具有正常的抑菌作用,但殺菌作用差。
{kind=link}
{kind=link}