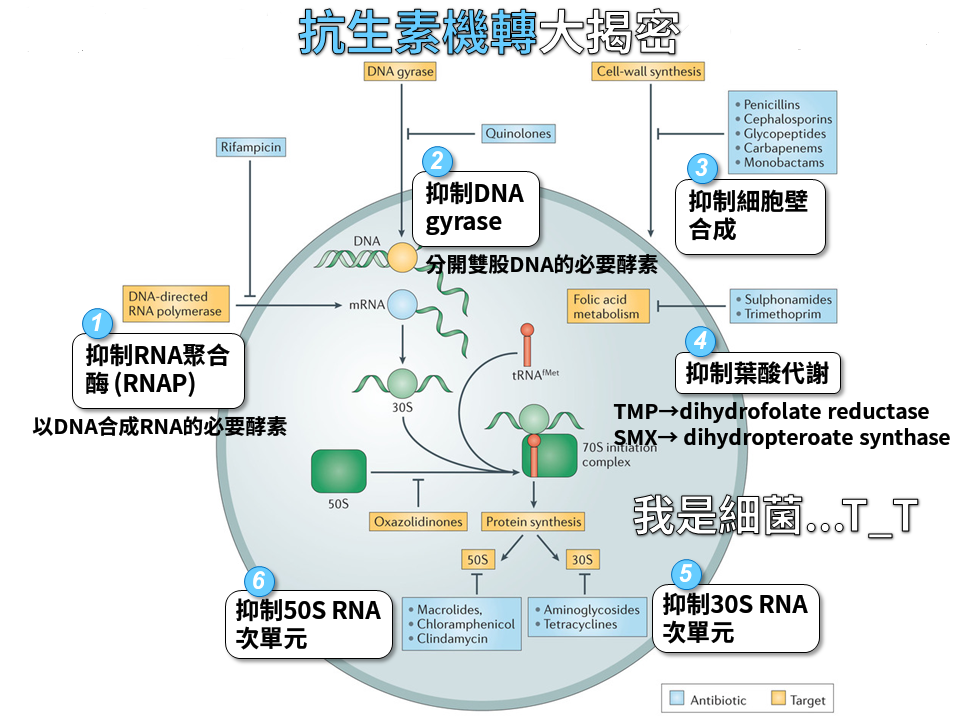
藥理學
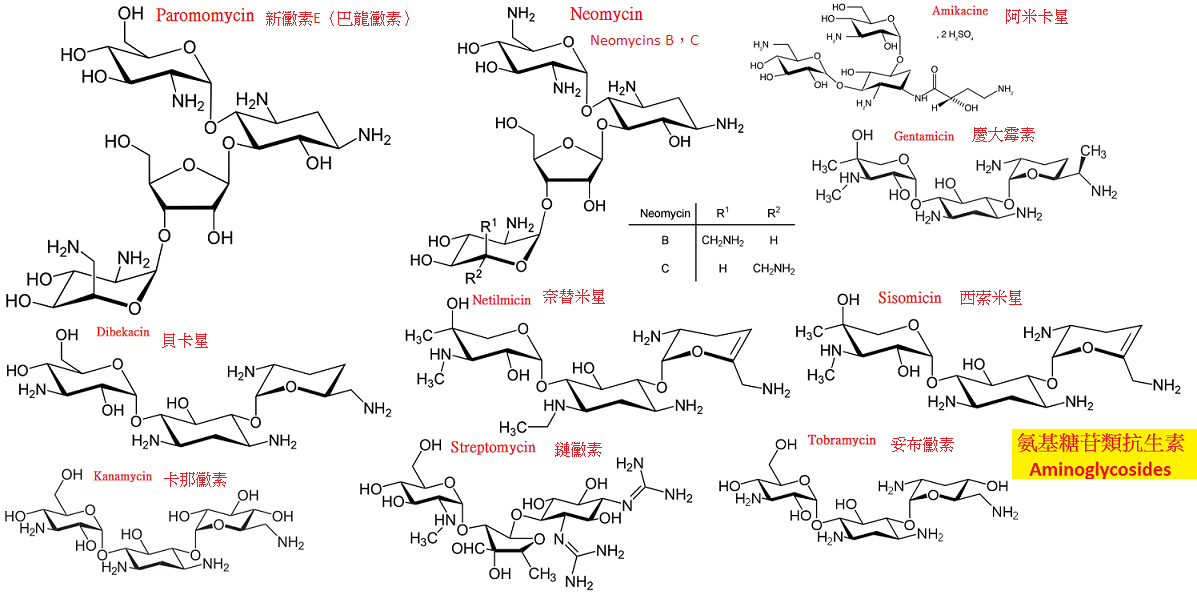
氨基甙類抗生素(aminoglycosides)都由氨基糖分子和非糖部分的甙元結合而成,
它包括鏈黴素、慶大黴素、卡那黴素、西索米星以及人工半合成的妥布黴素、阿米卡星、奈替米星等。
一、氨基甙類抗生素的共性
氨基甙類抗生素的化學結構基本相似,因此具有共同特點,如水溶性好,性質穩定;
此外,在抗菌譜,抗菌機制,血清蛋白結合率,胃腸吸收,經腎排泄,及不良反應等方面也有共性。
【抗菌作用】
氨基甙類對各種需氧革蘭陰性菌如大腸桿菌、克雷伯菌屬、腸桿菌屬、變形桿菌屬等具高度抗菌活性。
此外,對沙雷菌屬、產鹼桿菌屬、布氏桿菌、沙門菌、痢疾桿菌、嗜血桿菌及分枝桿菌也具有抗菌作用。
氨基甙類對革蘭陰性球菌如淋球菌、腦膜炎球菌的作用較差。
流感桿菌及肺炎支原體呈中度敏感,但臨床療效不顯著。
綠膿桿菌只對慶大黴素、阿米卡星、妥布黴素敏感,其中以妥布黴素為最強。
對各型鏈球菌的作用微弱,腸球菌對之多屬耐藥,但金葡菌包括耐青黴素菌株對之甚為敏感。
結核桿菌對鏈黴素、卡那黴素、阿米卡星和慶大黴素均敏感,但後者在治療劑量時不能達到有效抑菌濃度。
按相同重量比較,慶大黴素和西索米星的抗菌活性較卡那黴素、妥布黴素、奈替米星和阿米卡星稍強,
但臨床用量中它們的抗菌作用並無明顯差別。
【抗菌作用機制】氨基甙類的抗菌作用機制是阻礙細菌蛋白質的合成
。
許多基本成分如活化胺基酸,轉運核糖核酸(tRNA),
信息核糖核酸(mRNA),酶Mg2+,始動因子(F1,F2,F3),ATP,GTP等都參與了蛋白質合成(圖40-1)。
核蛋白體循環及有關抗生素作用部點陣圖解
圖40-1
核蛋白體循環及有關抗生素作用部點陣圖解
30s、50s表示組成核蛋白體的兩個亞基;A、P分別表示A位、P位,F1,F2,F3,為始動因子;
R表示終止因子;aa1、aa2、aa3-tRNA表示tRNA1、2、3
攜帶三種不同的活化胺基酸;
┗┷┛表示tRNA
上面三個小點表示反密碼,它能翻譯mRNA上相應的密碼;
mRNA上的1、2、3、T表示四個密碼,T為終止密碼,即停止合成的密碼,
T前可能有許多密碼,為圖示簡便,僅順序標出在T前的三個密碼。每個密碼包括三個核苷酸,稱為三聯密碼。
細菌蛋白質合成分為三個階段:
①起始階段 30S亞基與新生成的mRNA結合成mRNA-30S複合物,然後接上第一個氨基醯-tRNA(即甲醯蛋氨醯-tRNA),
接在相當於50S的P位,稱為30S起始複合物,後者很快與50S亞基結合成70S起始複合物。
②肽鏈延長階段
新的氨基醯-tRNA按mRNA的密碼要求進入核蛋白體50S亞基的A位上,
此時P位上的甲醯蛋氨醯或以後合成的肽鏈經肽醯轉移酶的作用,基羧基與A位新接上的胺基酸的氨基結合而形成肽鏈。
此時,在P位上的tRNA被釋放回到細胞質內轉運其他相應的胺基酸,
核蛋白體30S亞基上的mRNA發生移位,把帶有肽鏈的tRNA從A位移至P位。
空出的A位又接受新的氨基醯-tRNA,如此反覆使肽鏈不斷延長。
③終止階段mRNA上出現終止信號時,表示蛋白質合成已結束,
此時釋放因子(R)進入A位,使肽鏈釋放,tRNA及mRNA與核蛋白體分離,
核蛋白體70S又解離為30S與50S亞基,重新參與蛋白質合成。
氨基甙類能影響蛋白質合成的許多環節:
①起始階段,抑制70S始動複合物的形成,
②選擇性地與30S亞基上靶蛋白結合(如P10),使mRNA上的密碼錯譯,導致異常的、無功能的蛋白質合成;
③阻礙終止因子(R)與核蛋白體A位結合,使已合成的肽鏈不能釋放並阻止70S核蛋白體的解離,
最終造成菌體核心蛋白體的耗竭。
此外,氨基甙類通過離子吸附作用附著於細菌體表面造成胞膜缺損致使胞膜通透性增加,
細胞內鉀離子、腺嘌呤核苷酸、酶等重要物質外漏,從而導致細菌死亡。
氨基甙類與β內醯胺類都是殺菌藥,但它與後者不同,對靜止期細菌有較強的作用。
【體內過程】氨基甙類在胃腸道不吸收或極少吸收(<1%)。
口服後血藥濃度很低,可用於胃腸道消毒,但在腎功能損害時,
多次口服或直腸內給藥,血藥濃度可蓄積至中毒水平。肌內注射後氨基甙類吸收迅速且完全。
30∼90分鐘達到峰濃度。常用的氨基甙類藥代動力學參數見表40-1。
氨基甙類靜脈內給藥,其濃度高低隨劑量而異,一般在靜脈滴注20∼30分鐘後,
血漿中濃度與肌內注射者相同,本類藥物中除鏈黴素外,與血漿蛋白很少結合。
藥物主要分布於細胞外液,組織與細胞內藥物含量較低,分布容積大致與細胞外液容積相當,
成人為15L(0.56L/kg)。
腎臟皮質內藥物濃度可超過血藥濃度10∼50倍。消除t1/2平均可達112∼693小時。
腎臟皮質內藥物蓄積濃度越高,對腎毒性越大。
氨基甙類可進入內耳外淋巴液,濃度與用藥量成正比,其t1/2較血漿t1/2長5∼6倍(圖40-2)。
當腎功能減退(無尿)時其濃度與t1/2均明顯增加。
氨基甙類在體內不被代謝,約90%以原形經腎小球過濾排出,尿藥濃度極高,約為血漿峰濃度的25∼100倍。
表40-1 常用氨基甙類的藥代動力學
抗生素 IM.
血藥濃度達
峰時間(小時)
t1/2(小時) 24小時尿
排出(%)
蛋白結合率(%)
正常
無尿
鏈黴素 0.5∼1.5 2.0∼3.0 50∼110 80
35
慶大黴素 0.75∼1.0
1.7∼2.3 48∼72 70∼80
<10
妥布黴素 0.33∼0.75
2.0∼2.8 56∼60 80∼90
<10
卡那黴素 0.75∼1.0
2.1∼2.4 60∼90 84∼90
0
阿米卡星 0.75∼2.0
2.2∼2.5 56∼150 81∼98 4.0
西索米星 0.75∼1.0
2.0∼2.3 35∼37 85∼87
0
奈替米星 0.5∼1.0
2.2 33
80∼90
<10
正常和無尿豚鼠慶大黴素皮下注射50mg/kg後
圖40-2
正常和無尿豚鼠慶大黴素皮下注射50mg/kg後血清(A)和外淋巴液(B)的藥動學
慶大黴素濃度用生物測定法,每點為6隻動物的均值,無尿動物於給藥前3小時切除腎臟
【不良反應】
1.耳毒性
臨床反應可分為二類:一為前庭功能損害,有眩暈、噁心、嘔吐、眼球震顫和平衡障礙,其發生率依次為:
新黴素(已少用)>卡那黴素>鏈黴素>西索米星>慶大黴素>妥布黴素>奈替米星。
另一為耳蝸神經損害,表現為聽力減退或耳聾,其發生率依次為:
新黴素>卡那黴素>阿米卡星>西索米星>慶大黴素>妥布黴素>鏈黴素。
必須指出耳聾性的許多自覺症状並不明顯,但經儀器監測顯示有前庭功能
或聽力損害的「亞臨床耳毒性」反應的發生率則可達10%∼20%,最先影響為高頻聽力,隨後逐漸波及低頻部分。
耳毒性發生機制可能是內耳淋巴液中藥物濃度過高,損害內耳柯蒂氏器內、
外毛細胞的糖代謝和能量利用,導致內耳毛細胞膜上鉀鈉離子泵發生障礙,終使毛細胞的功能受損。
為防止和減少耳毒性反應,在治療過程中應注意觀察耳鳴、眩暈等早期症状的出現,
進行聽力監測,並根據患者的腎功能(肌酐清除率等)及血藥濃度來調整用藥劑量。
除非必要,應避免與高效利尿藥或其他耳毒性藥物合用。
2.腎毒性
氨基甙類主要經腎排泄並在腎(尤其是皮質部)蓄積,主要損害近曲小管上皮細胞,
但不影響腎小球,臨床化驗可見蛋白尿、管形尿、尿中紅細胞、腎小球過濾減少,嚴重者可發生氮質血症及無尿等。
年老、劑量過高以及與其他腎毒性藥物如呋塞米、多粘菌素、兩性黴素B等合用時容易發生腎功能損害,
在常用劑量時各藥對腎的毒性順序為:新黴素>卡那黴素>妥布黴素>鏈黴素,奈替米星腎毒性很低。
3.神經肌肉阻斷作用
各種氨基甙類抗生素均可引起神經肌肉麻痹作用,雖較小見,但有潛在性危險。
神經肌肉阻斷作用與劑量及給藥途徑有關,如靜脈滴注速度過快或同時應用肌肉鬆弛劑與全身麻醉藥。
重症肌無力者尤易發生,可致呼吸停止。
其機制是乙醯膽鹼的釋放需Ca2+的參與,藥物能與突觸前膜上「鈣結合部位」結合,
從而阻止乙醯膽鹼釋放。當出現神經肌肉麻痹時,可用鈣劑或新斯的明治療。
4.過敏反應
氨基甙類可以引起嗜酸粒細胞增多,各種皮疹,發熱等過敏症状,也可引起嚴重過敏休克,
尤其是鏈黴素引起的過敏休克發生率僅次於青黴素G,應引起警惕。
二、各種氨基甙類抗生素藥理特點及應用
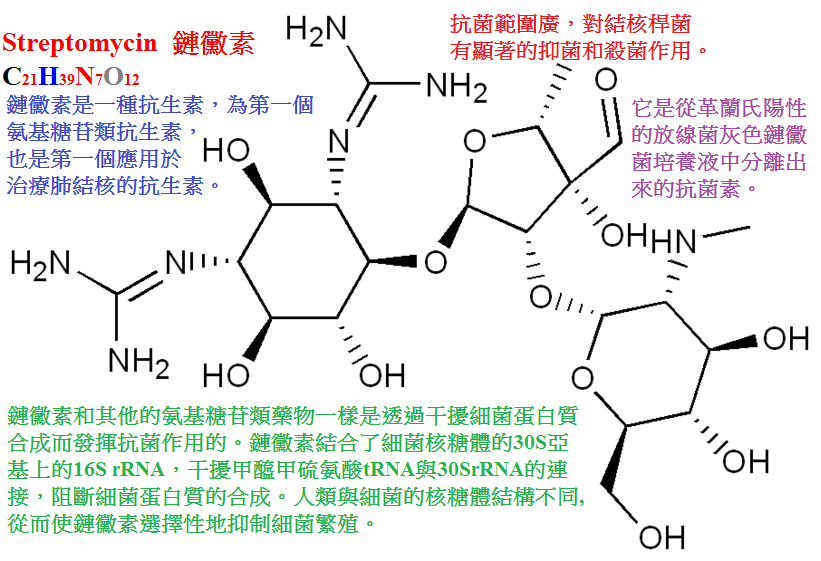
1.鏈黴素(streptomycin)是由鏈絲菌培養液提取而得,常用其硫酸鹽,性質穩定,水溶液在室溫可保持一周。
口服不吸收,肌肉注射吸收快,30∼60分鐘達峰濃度,t1/2為2∼3小時,一次注射有效濃度可達6∼8小時,
年齡超過40歲t1/2可延長至9小時,主要分布於細胞外液,大部分經腎排泄,腎功不全時,排泄減慢。
鏈黴素對多數革蘭陰性菌有強大抗菌作用,但因毒性與耐藥性問題,限制了它的臨床應用。
目前臨床主要用於:
①鼠疫與兔熱病,對此鏈黴素是首選藥;
②布氏桿菌病,鏈黴素與四環素合用也有滿意的效果;
③感染性心內膜炎,對草綠色鏈球菌引起者,以青黴素合併鏈黴素為首選;
對腸球菌引起者,也需青、鏈合用治療,但部分菌株對鏈黴素耐藥,可改用慶大黴素或妥布黴素等;
④結核病,鏈黴素為最早的抗結核藥,現仍有應用,但必須與其他抗結核藥聯合應用,以延緩耐藥性的發生;
⑤鏈黴素與青黴素或氨苄西林合用,可用於預防常發的細菌性心內膜炎及呼吸、胃腸道及泌尿系統手術後感染。
鏈黴素治療時常可出現頭痛、頭暈、嘔吐、耳鳴、平衡失調和眼球震顫。多是可逆的。
嚴重者可致永久性耳聾。對腎臟的毒性為氨基甙類中最輕者,但腎功能不全者仍應慎用。
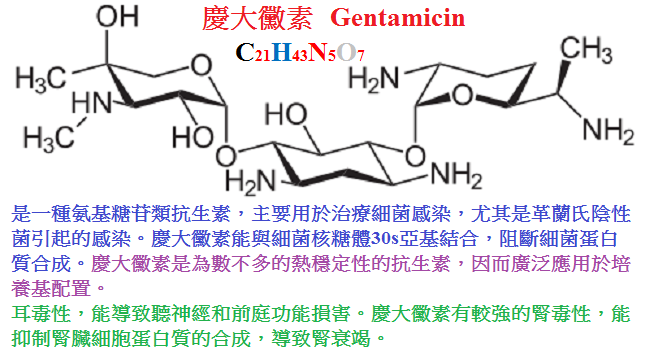
2.慶大黴素(gentamicin)是目前臨床最為常用的廣譜氨基甙類。
慶大黴素水溶液穩定,水針劑常作肌內或靜脈滴注給藥。體內過程與鏈黴素相仿(表40∼1)。
但其有效與安全的血藥濃度較低(4∼8mg/L)。
藥物主要經腎排泄,部分經膽汁入腸,膽汁藥物濃度可達血藥濃度的60%∼80%,t1/2約3小時。
慶大黴素廣泛用於治療敏感菌的感染:
①嚴重革蘭陰性桿菌的感染如敗血症、骨髓炎、肺炎、腹膜感染、腦膜炎等、慶大黴素是首選藥;
②綠膿桿菌感染,慶大黴素常與羧苄西林合用可獲協同作用,但兩藥不可同時混合滴注,因後者可使本藥的活力降低;
③病因未明的革蘭陰性桿菌混合感染,慶大黴素與廣譜半合成青黴素類(羧苄西林或哌拉西林等)或頭孢菌素聯合應用可以提高療效;
④與青黴素聯合治療腸球菌心內膜炎;與羧苄西林、氯黴素聯合治療革蘭陰性桿菌心內膜炎;
⑤慶大黴素口服可用於腸道感染或腸道術前準備;
⑥慶大黴素局部用於皮膚、粘膜表面感染、眼、耳、鼻部感染,但因可致光敏感反應,大面積應用易致吸收毒性,
故少作局部應用。 不良反應有前庭神經功能損害,但較鏈黴素少見,對腎臟毒性則較多見。
3.卡那黴素(kanamycin)由鏈絲菌培養液獲得。
卡那黴素體內過程與鏈黴素、慶大黴素基本相同。其抗菌譜與鏈黴素相似,但稍強,對多數常見的革蘭陰性菌及結核菌有效,
但對綠膿桿菌無效。體內抗菌有效的血藥濃度範圍為8∼16µg/ml。
卡那黴素由於毒性及耐藥菌較多見,其在臨床應用已為慶大黴素等其他氨基甙類藥所取代。
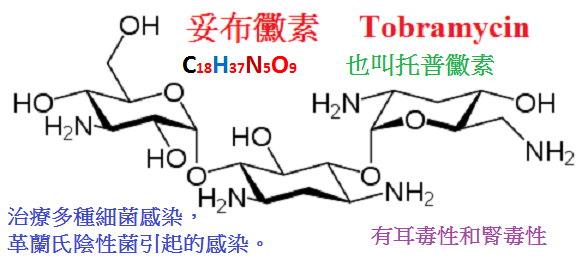
4.妥布黴素(tobramycin)由鏈絲菌培養液中提得,也可由卡那黴素B脫氧而成,其水溶液非常穩定。
抗菌作用與慶大黴素相似,對絕大多數腸桿菌科細菌、綠膿桿菌及葡萄球菌具良好的抗菌作用。
最突出的是對綠膿桿菌作用較慶大黴素強2∼4倍,並且對慶大黴素耐藥者仍有效,對腸球菌及除綠膿桿菌外的假單孢菌屬及厭氧菌無效,
對肺炎桿菌、腸桿菌屬與變形桿菌屬的作用較慶大黴素略強;但對沙雷菌和沙門菌的作用略差。
妥布黴素與慶大黴素相同,主要用於各種嚴重的革蘭陰性桿菌感染,但一般不作為首選藥。
對綠膿桿菌感染或需較長時間用藥者,如感染性心內膜炎,以選用妥布黴素為宜。
妥布黴素的耳毒性較慶大黴素略低,但仍應警惕。一般每日劑量不宜超過5mg/kg,血藥濃度不宜超過12mg/L。
在腎功能減退時還應根據血清肌酐清除率,調整劑量與給藥間隔。
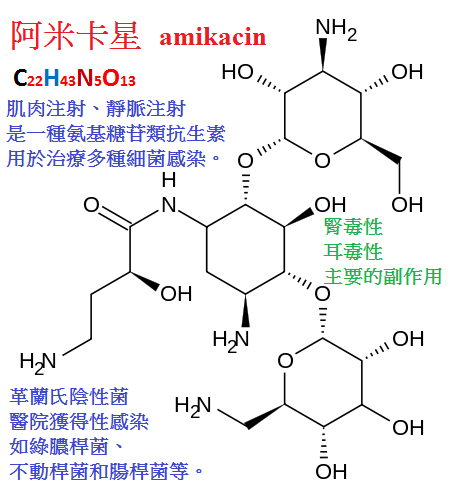
5.阿米卡星(amikacin,丁胺卡那黴素)是卡那黴素的半合成衍生物,
其抗菌譜為本類藥物中最寬的。其突出優點是對許多腸道革蘭陰性菌和綠膿桿菌所產生的鈍化酶穩定,
因而主要用於治療對其他氨基甙類耐藥菌株(包括綠膿桿菌)所致的感染,
如對慶大黴素、卡那黴素耐藥株引起的尿路、肺部感染,以及綠膿、變形桿菌所致的敗血症。
與羧苄西林或頭孢噻吩合用。連續靜脈滴注治療中性粒細胞減少或其他免疫缺陷者感染,可獲得滿意效果。
阿米卡星僅可為革蘭陰性菌所產生的一種乙醯轉移酶AAC(b')所鈍化而耐藥,
此外,由於細胞壁屏障作用,致使藥物不能有效滲入細菌體也可導致耐藥株產生。
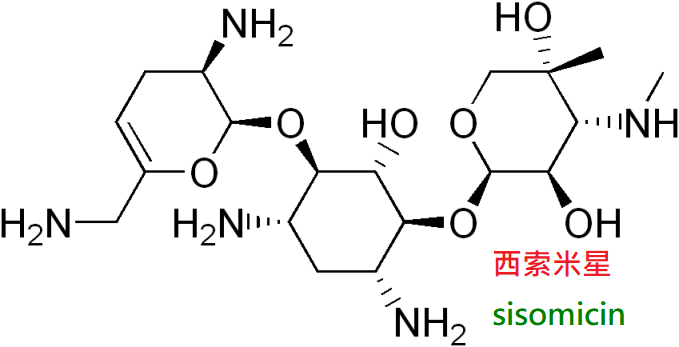
6.西索米星(sisomicin)由小單孢菌發酵液中獲得,藥用其硫酸鹽,易溶於水。
抗菌譜及體內過程與慶大黴素很相似,抗綠膿桿菌作用比慶大黴素強兩倍,
對金葡菌、克雷伯菌屬、球菌屬、大腸桿菌、變形桿菌和化膿性球菌也有良效。
臨床上用於上述細菌引起的感染。毒性約比慶大黴素大兩倍。
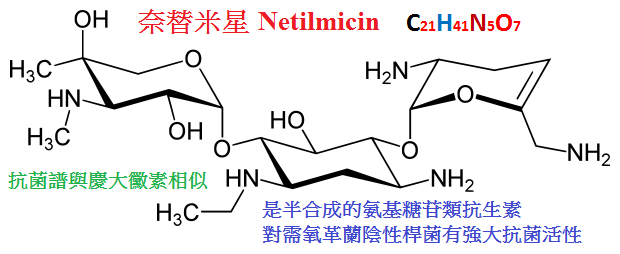
7.奈替米星(netilmicin)是新的氨基甙類抗生素。
其藥動學特性與慶大黴素、妥布黴素相似,它也像阿米卡星不被大多數鈍化酶滅活。
對一些革蘭陰性桿菌,如大腸桿菌、克雷伯桿菌、沙雷桿菌、各型變形桿菌和綠膿桿菌都具有較強抗菌活性,
對流感嗜血桿菌、沙門菌、志賀菌和奈瑟菌也有效。
對某些耐其他氨基甙類的革蘭陰性桿菌及耐青黴素類的金葡菌也有效。
適用於尿路、腸道、呼吸道、皮膚軟組織、骨和關節、腹腔及創口部分的感染。
奈替米星的耳、腎毒性是氨基甙類抗生素中最低者。但仍宜注意。
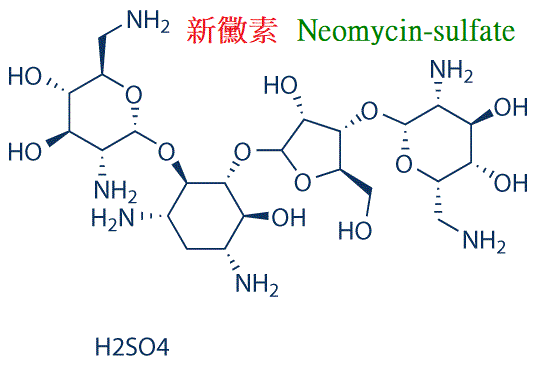
8.新黴素(neomycin)抗菌譜似卡那黴素。
肌內注射吸收快,毒性比卡那黴素大,易引起永久性耳聾和腎損害,不宜注射給藥。
口服很少吸收,毒性較小,可用於腸道感染和腸道消毒。局部外用治療皮膚粘膜淺表感染。
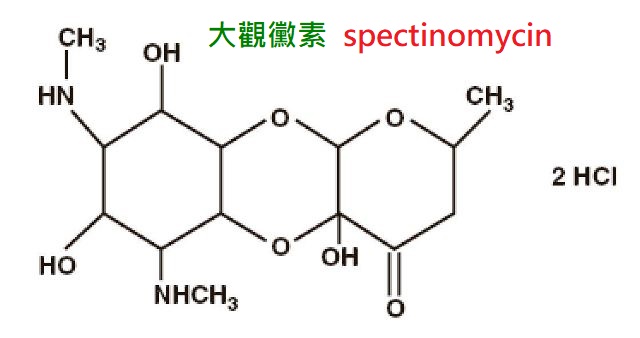
【附】:大觀黴素(spectinomycin)由鏈黴菌所產生的一種氨基環醇類(aminocyclitols)抗生素,
主要對淋球菌有高度抗菌活性,6.3mg/L可抑制大多數淋球菌。
肌注2g,1小時血藥濃度達峰100mg/L,t1/2約2.5小時。
藥物主要經尿排泄。臨床的唯一適應證是無併發症的淋病,限於對青黴素、四環素等耐藥的淋病或患者對青黴素過敏者。
三、藥物相互作用
氨基甙類與兩性黴素、桿菌肽、頭孢噻吩、多粘菌素或萬古黴素合用能增加腎臟毒性。
呋塞米(速尿)、利尿酸及甘露醇等能增加氨基甙類的耳毒性。
苯海拉明、敏可靜、安其敏等抗組胺藥可掩蓋氨基甙類的耳毒性。
氨基甙類能增強骨骼肌鬆弛藥及全身麻醉藥引起的肌肉鬆弛作用,可導致呼吸抑制。
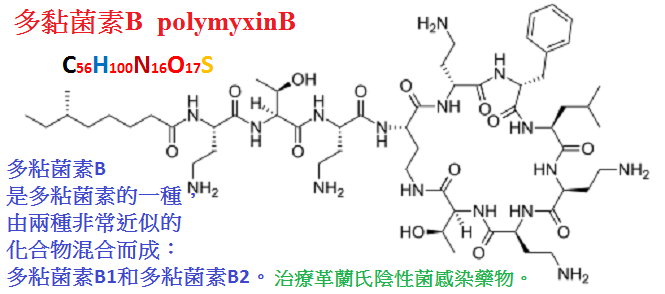
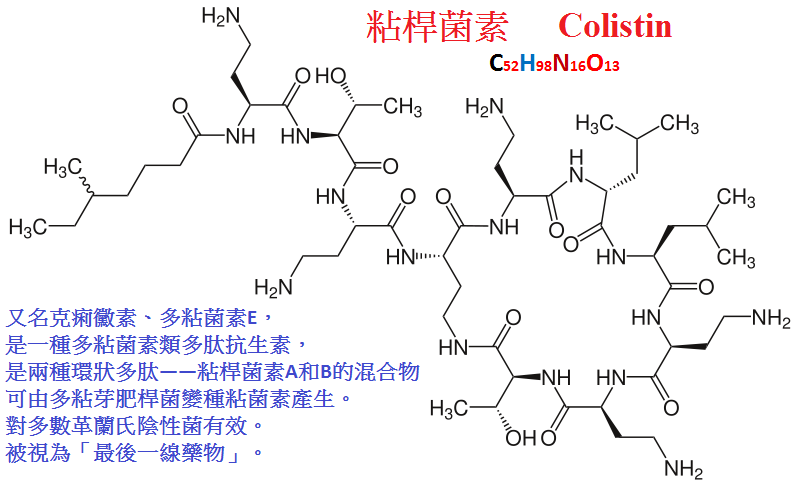
多粘菌素包括多粘菌素B(polymyxin
B)及多粘菌素E(polymyxin
E;粘菌素,colistin),
二者具有相似的藥理作用。是多肽類抗生素,由於靜脈給藥可致嚴重腎毒性現已少用。
【體內過程】多粘菌素口服不易吸收。肌內注射50mg後2小時血藥濃度達峰值(2∼8mg/L),
有效血藥濃度可維持8∼12小時,t1/2約6小時。腎功能不全者清除慢,t1/2可達2∼3天。
它分布於全身組織,以肝、腎為最高,並保持較長時間。
多粘菌素不易彌散進入胸、腹腔、關節腔,即使在腦膜炎症時也不易透入腦脊液中,膽汁中濃度也較低。
藥物經腎緩慢排泄。
【抗菌作用及臨床應用】對多數革蘭陰性桿菌有殺滅作用。
多肽類抗生素具有表面活性,含有帶陽電荷的游離氨基,能與革蘭陰性菌細胞膜的磷脂中帶陰電荷的磷酸根結合,
使細菌細胞膜面積擴大,通透性增加,細胞內的磷酸鹽、核苷酸等成份外漏,導致細菌死亡。
多粘菌素對生長繁殖期和靜止期的細菌都有效,
過去曾用於對其他抗生素耐藥的綠膿桿菌和革蘭陰性桿菌所致感染如敗血症、腦膜炎、心內膜炎、燒傷後感染等。
但現在已被療效好、毒性低的其他抗生素所取代。
仍可局部用於敏感菌的眼、耳、皮膚、粘膜感染及燒傷綠膿桿菌感染。多粘菌素口服用於腸道手術前準備。
【不良反應】毒性較大。主要表現在腎臟及神經系統兩方面,其中多粘菌素B較E尤為多見,
症状為蛋白尿、血尿等。大劑量、快速靜脈滴注時,由於神經肌肉的阻滯可導致呼吸抑制。
硫酸鏈黴素(streptomycin
sulfate)
硫酸慶大黴素(gentamicin
sulfate)
硫酸卡那黴素(kanamycin
sulfate)
硫酸妥布黴素(tobramycin
sulfate)
硫酸阿米卡星(amikacin
sulfate)
硫酸西索米星(sisomicin
sulfate)
硫酸奈替米星(netilimicin
sulfate)
硫酸新黴素(neomycin
sulfate)
大觀黴素(spectinomycin)
硫酸多粘菌素B(polymyxin
B sulfate)
硫酸多粘菌素E(polymyxin
E sulfate,colistin
sulfate)
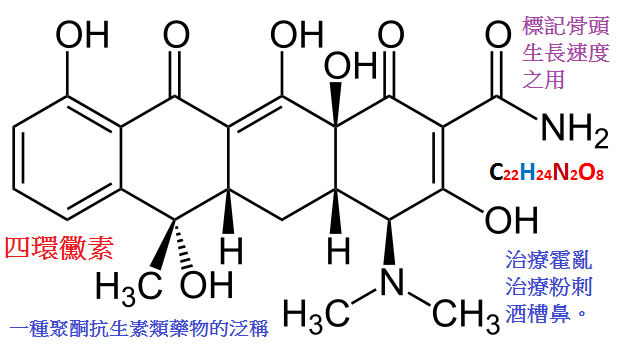
四環素類和氯黴素的抗菌譜極廣,包括革蘭氏陽性和陰性菌、立克次體、衣原體、支原體和螺旋體,故常稱為廣譜抗生素。
四環素類抗生素具有共同的基本母核(氫化駢四苯),僅取代基有所不同。
它們是兩性物質,可與鹼或酸結合成鹽,在鹼性水溶液中易降解,在酸性水溶液中則較穩定,故臨床一般用其鹽酸鹽。
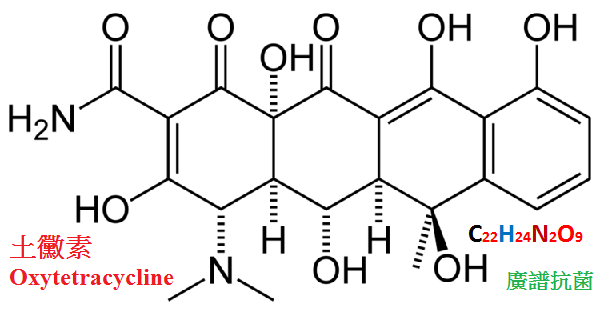
四環素類可分為天然品與半合成品兩類。天然品有金黴素、土霉素、四環素和去甲金黴素等。
金黴素已被淘汰,去甲金黴素我國不生產。四環素和土霉素較常用。
半合成品有多西環素和米諾環素,前者在我國較為常用。
四環素(tetracycline)和土霉素(氧四環素,oxytetracycline),由於抗菌譜廣,口服有效,應用方便,故曾長期廣用於臨床。
近年來由於耐藥菌株日益增多,療效不夠理想,且副作用較多,其臨床應用已明顯減少。
【抗菌作用】抗菌譜廣,對革蘭陽性的肺炎球菌、溶血性鏈球菌、草綠色鏈球菌及部分葡萄球菌、破傷風桿菌和炭疽桿菌等;
對革蘭陰性細菌中的腦膜炎球菌、痢疾桿菌、大腸桿菌、流感桿菌、巴氏桿菌屬、布氏桿菌等及某些厭氧菌
(如擬桿菌、梭形桿菌、放線菌)都有效。此外,對肺炎支原體、立克次體、螺旋體、
放線菌也有抑制作用,還能間接抑制阿米巴原蟲。對綠膿桿菌、病毒與真菌無效。
四環素類屬快速抑菌劑,在高濃度時也有殺菌作用。
其抗菌機制主要為與細菌核蛋白體30S亞單位在A位特異性結合,阻止aa-tRNA在該位置上的聯結,
從而阻止肽鏈延伸和細菌蛋白質合成。
其次四環素類還可引起細胞膜通透性改變,使胞內的核苷酸和其他重要成分外漏,從而抑制DNA複製。
細菌對四環素類的耐藥性在體外發展較慢,然本類藥物之間有交叉耐藥性。
大腸桿菌和其他腸桿菌科細菌的耐藥性主要通過耐藥質體介導,並可傳遞、
誘導其他敏感細菌轉成耐藥,帶耐藥質體細菌的細胞膜對四環素類藥物攝入減少或泵出增加。
【體內過程】口服易吸收,但不完全,四環素吸收較土霉素好,
2∼4小時血藥濃度可達高峰,t1/2約為8.5小時,土霉素血藥濃度較低,t1/2為9.6小時,
由於四環素類能與多價陽離子如Mg2+、Ca2+、Al3+及Fe2+等起絡合作用,
因而含這些離子的藥物和食物均可妨礙其吸收。
飯後服鹽酸四環素較空腹服用時血藥濃度低50%左右;鐵劑可使四環素的吸收率下降約40%∼90%,
如需要兩藥合用,服藥時間應相隔3小時。胃液中酸度高時,藥物溶解完全,吸收較好。
此外,口服四環素與土霉素吸收量有一定限度。
服藥量超過0.5g以上,血藥濃度並不隨劑量增加而提高,只增加糞便中的排泄量。
吸收後廣泛分布於各組織中,並能沉積於骨及牙組織內。
它們與血漿蛋白結合率約為20%∼30%,因此四環素容易滲入胸腔、腹腔、胎兒循環及乳汁中,
但不易透過血腦屏障,腦脊液中的藥物濃度一般僅為血藥濃度的1/10。
四環素、土霉素主要以原形經腎小球過濾排出,故尿藥濃度較高,有利於治療尿路感染。
土霉素口服排泄快,且較完全,排泄量可達60%∼70%。四環素排泄量較少,約20%∼30%。
本類藥物經肝濃縮排入膽汁,形成肝腸循環。膽汁中藥物濃度為血藥濃度的10∼20倍。
【臨床應用】四環素類臨床應用範圍比較廣泛。
對立克次體感染和斑疹傷寒、恙蟲病以及支原體引起的肺炎有良效,為首選藥物。
對革蘭陽性菌和陰性菌感染,百日咳、痢疾、肺炎桿菌所致的尿道、呼吸道與膽道感染,可用新四環素類作次選藥。
【不良反應】
1.胃腸道反應
本藥口服後直接刺激而引起噁心、嘔吐、上腹不適、腹脹、腹瀉等症状,尤以土霉素多見,與食物同服可以減輕。
2.二重感染 正常人的口腔、鼻咽、腸道等都有微生物寄生,菌群間維持平衡的共生狀態。
廣譜抗生素長期應用,使敏感菌受到抑制,而不敏感菌乘機在體內繁殖生長,造成二重感染,又稱菌群交替症。
多見於老幼和體質衰弱、抵抗力低的患者。
此外,合併應用腎上腺皮質激素、抗代謝或抗腫瘤藥物也更容易誘發二重感染。
常見的二重感染有:
①真菌病,致病菌以白色念珠菌最多見。表現為口腔鵝口瘡、腸炎、可用抗真菌藥治療。
②葡萄球菌引起的假膜性腸炎,此時葡萄球菌產生強烈的外毒素,引起腸壁壞死,體液滲出、
劇烈腹瀉、導致失水或休克等症状,有死亡危險。此種情況必須停藥並口服萬古黴素。
3.對骨、牙生長的影響 四環素類能與新形成的骨、牙中所沉積的鈣相結合。
妊娠五個月以上的婦女服用這類抗生素,可使出生的幼兒乳牙釉質發育不全並出現黃色沉積,引起畸形或生長抑制。
4.其他 長期大量口服或靜脈給予(每日超過1∼2g)可造成嚴重肝臟損害。
也能加劇原有的腎功能不全,影響胺基酸代謝而增加氮質血症。
此外,四環素類抗生素還可引起藥熱和皮疹等過敏反應。
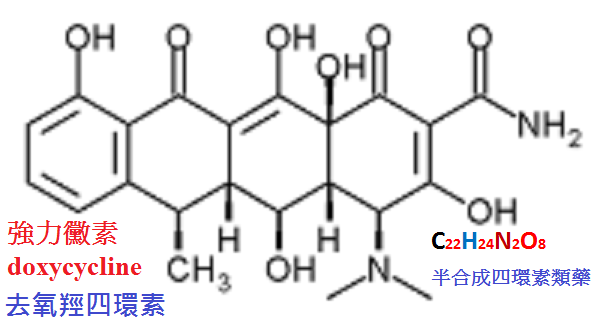
多西環素(doxycycline,強力黴素)是土霉素的脫氧物。易溶,遇光不穩定。
【抗菌作用】抗菌譜和四環素相似。但抗菌作用強2∼10倍,且對土霉素、四環素的耐藥金葡菌有效。
【體內過程】脂溶性較大,因此口服吸收快而完全,分布於全身,腦脊液中濃度也較高。
強力黴素的吸收不受食物的影響。藥物大部經膽汁排入腸道又可再吸收,經腎小管時也可再吸收,
因此t1/2長達20小時,可維持有效血藥濃度24小時以上。
一般細菌性感染每日服藥一次即可。藥物小部分從腎排泄。
大部分以結合或絡合的無活性代謝產物由糞便排泄,故對腸道菌群無影響,腎功能不全時仍可使用。
【臨床應用】同四環素,用於呼吸道感染如老年慢性氣管炎、肺炎、麻疹肺炎,
也用於泌尿道感染及膽道感染等。對腎功能不良患者的腎外感染也可使用。
對產腸毒素大腸桿菌所致的腹瀉也有效,但宜慎用。
【不良反應】常見胃腸道刺激性反應,如噁心、嘔吐、腹瀉、舌炎、口腔炎及肛門炎等,宜飯後服藥。
皮疹及二重感染少見。在靜脈注射過程中可出現舌頭麻木及口內特殊氣味,個別可有嘔吐。
【相互作用】多西環素與肝藥酶誘導劑苯巴比妥、苯妥英鈉等同服,
可使其t1/2縮短為7小時左右,並使血藥濃度降低而影響療效。
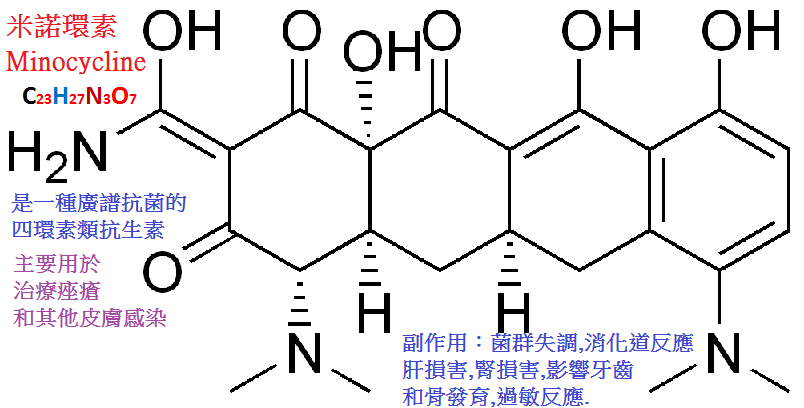
米諾環素(minocycline,二甲胺四環素)是長效高效的半合成四環素,
其抗菌譜和四環素相近,抗菌作用為四環素類中最強,對四環素耐藥的金葡菌、鏈球菌和大腸桿菌對本品仍敏感。
口服吸收迅速,2∼3小時後血藥濃度可達高峰,經尿與糞排泄量為本類藥中最低者,
t1/2約為13(10∼20)小時。藥物在體內長時間存留於脂肪組織,給藥後10天尿中仍可測出。
臨床用於尿路、胃腸道、呼吸道感染、膿皮病、骨髓炎,眼耳鼻喉部感染等。此外對瘧疾也有一定效果。
不良反應與其他四環類基本相同,但能引起可逆性前庭反應,
包括噁心、嘔吐、頭昏、眼花及運動失調等,常在開始服藥時出現,停藥後24∼48小時可消失。
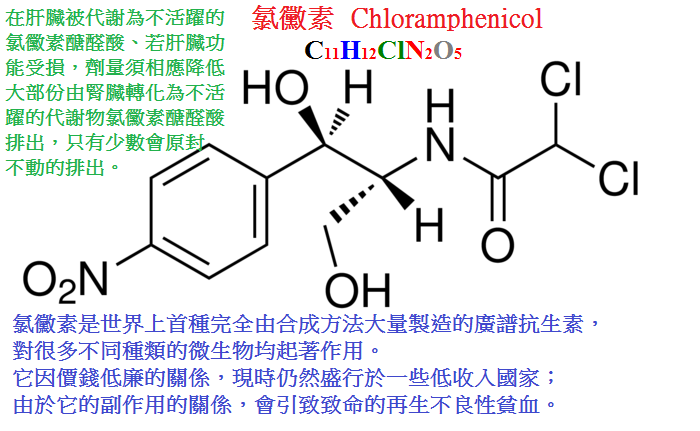
氯黴素(chloramphenicol,chloromycetin)是由委內瑞拉鏈絲菌產生的抗生素。分子中含有氯。
【抗菌作用】氯黴素對革蘭陽性、陰性細菌均有抑制作用,且對後者的作用較強。
其中對傷寒桿菌、流感桿菌、副流感桿菌和百日咳桿菌的作用比其他抗生素強,
對立克次體感染如斑疹傷寒也有效,但對革蘭陽性球菌的作用不及青黴素和四環素。
抗菌作用機制是與核蛋白體50S亞基結合,抑制肽醯基轉移酶,從而抑制蛋白質合成。
各種細菌都能對氯黴素發生耐藥性,其中以大腸桿菌、痢疾桿菌、變形桿菌等較為多見,傷寒桿菌及葡萄球菌較少見。
細菌對氯黴素產生耐藥性比較慢,可能是通過基因的逐步突變而產生的,但可自動消失。
細菌也可以通過R因子的轉移而獲得耐藥性,獲得R因子的細菌能產生氯黴素乙醯轉移酶(acetyltransferase)使氯黴素滅活。
【體內過程】氯黴素自腸道上部吸收,一次口服1.0g後2小時左右血中藥物濃度可達到峰值(約10∼13mg/L)。
血漿t1/2平均為2.5小時,6∼8小時後仍然維持有效血藥濃度。
氯黴素廣泛分布於各組織和體液中,腦脊液中的濃度較其他抗生素為高。
氯黴素的溶解和吸收均與製劑的顆粒大小及晶型有關。
肌內注射吸收較慢,血濃度較低,僅為口服同劑量的50%∼70%,但維持時間較長。
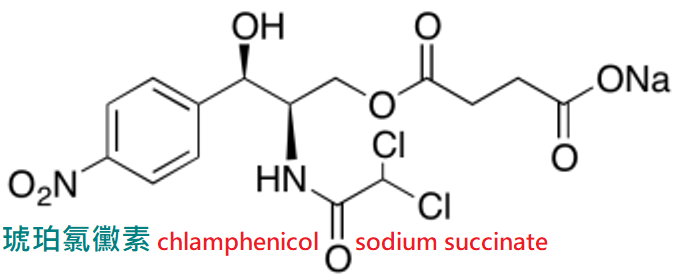
注射用氯黴素為琥珀酸鈉鹽,水中溶解度大,在組織內水解產生氯黴素。
氯黴素在體內代謝大部分是與葡萄糖醛酸相結合,其原形藥及代謝物迅速經尿排出,
口服量5%∼15%的有效原形藥經腎小球過濾而排入尿中,並能達到有效抗菌濃度,可用於治療泌尿系統感染。
腎功能不良者使用時應減量。
【臨床應用】氯黴素曾廣泛用於治療各種敏感菌感染,後因對造血系統有嚴重不良反應,
故對其臨床應用現已做出嚴格控制。可用於有特效作用的傷寒、副傷寒和立克次體病等及敏感菌所致的嚴重感染。
氯黴素在腦脊液中濃度較高,也常用於治療其他藥物療效較差的腦膜炎患者。必要時可用靜脈滴注給藥。
【不良反應】主要不良反應是抑制骨髓造血機能。
症状有二:一為可逆的各類血細胞減少,其中粒細胞首先下降,這一反應與劑量和療程有關。
一旦發現,應及時停藥,可以恢復;二是不可逆的再生障礙性貧血,雖然少見,但死亡率高。
此反應屬於變態反應與劑量療程無直接關係。
可能與氯黴素抑制骨髓造血細胞內粒線體中的與細菌相同的70S核蛋白體有關。
為了防止造血系統的毒性反應,應避免濫用,應用時應勤查血象,氯黴素也可產生胃腸道反應和二重感染。
此外,少數患者可出現皮疹及血管神經性水腫等過敏反應,但都比較輕微。
新生兒與早產兒劑量過大可發生循環衰竭(灰嬰症候群),這是由於他們的肝發育不全,排泄能力差,使氯黴素的代謝、
解毒過程受限制,導致藥物在體內蓄積。因此,早產兒及出生兩周以下新生兒應避免使用。
【應用注意】
1.開始治療前應檢查血象(白細胞、分類及網織細胞計數),隨後每48小時再查一次,
治療結束還要定期檢查血象,一旦出現異常,應立即停藥。
2.氯黴素治療時,對用口服降血糖藥的糖尿病患者或服抗凝血藥者,尤其是老年人,
應分別檢測血糖及凝血酶原時間,以防藥效及毒性增強。
3.對肝腎功能不良,G-6PDH缺陷者、嬰兒、孕婦、乳婦應慎用。
4.用藥時間不宜過長,一般不超過二個月,能達到防止感染複發即可,避免重複療程。
藥理學/四環素類及氯黴素製劑與用法
鹽酸四環素(tetracycline hydrochloride)
鹽酸土霉素(oxytetracycline hydrochloride)
多西環素(doxycycline)
米諾環素(minocycline)
氯黴素(chloramphenicol,chloromycetin)
琥珀氯黴素(chlamphenicol
sodium succinate)
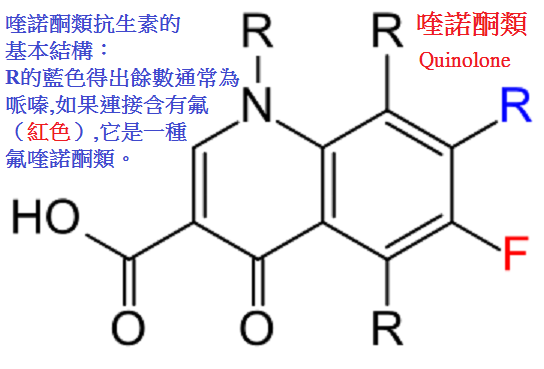
喹諾酮類quinolones是人工合成的含4-喹諾酮基本結構,對細菌DNA螺旋酶(DNA
gyrase)
具有選擇性抑制作用的抗菌藥物。目前發展迅速,臨床廣為使用。
一、喹諾酮類藥物概述
(一)簡史
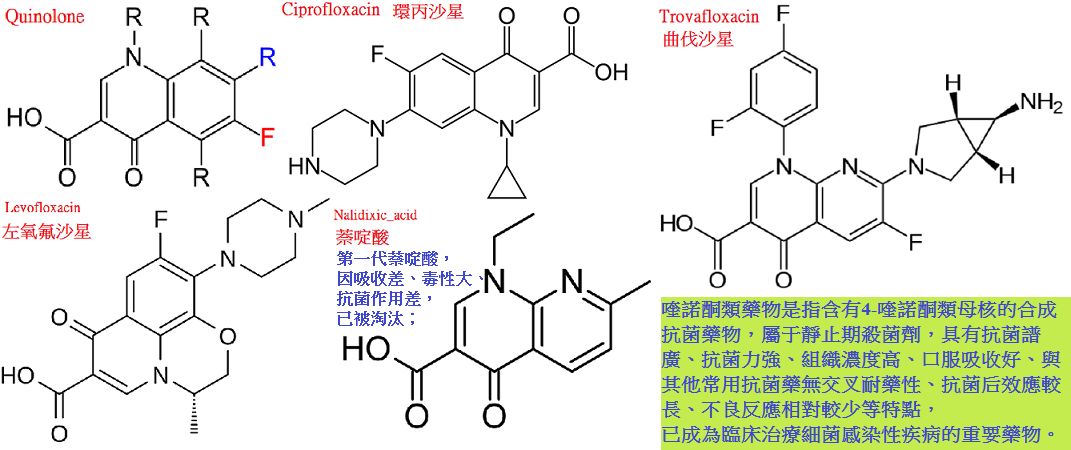
萘啶酸(nalidixicacid)是1962年用於臨床的第一個喹諾酮類藥(實是萘啶酮),
抗菌譜窄,口服吸收差,副作用多,現已不用。
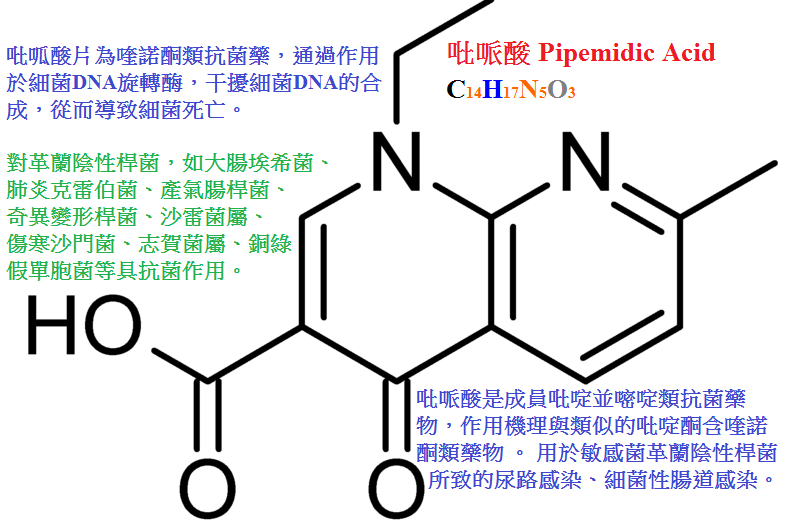
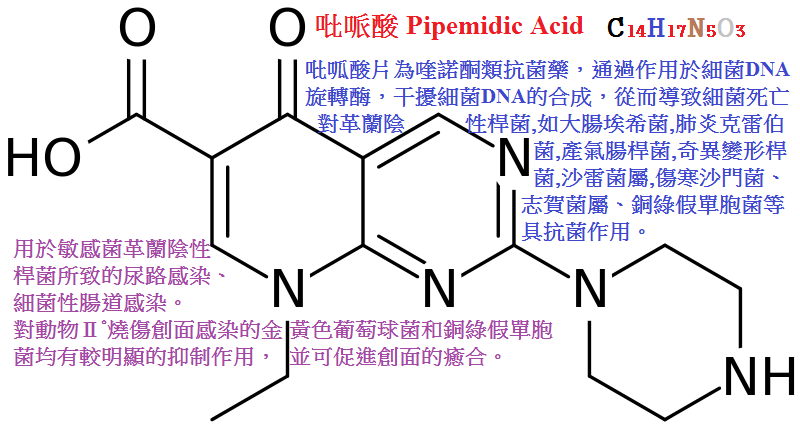
吡哌酸(pipemidicacid)抗菌活性強於萘啶酸,口服少量吸收,不良反應較萘啶酸少,可用于敏感菌的尿路感染與腸道感染。
1979年合成諾氟沙星(norfloxacin),隨又合成一系列含氟的新喹諾酮類藥,通稱為氟喹諾酮類(fluoroquinolones)。
(二)化學結構與作用關係
本類藥物的構效關係研究表明:4-喹諾酮母核的3位均有羧酸基,6位引入氟原子可增強抗菌作用並對金葡菌有抗菌活性;
7位引進哌嗪環可提高對金葡菌及綠膿桿菌的抗菌作用(如諾氟沙星),哌嗪環被甲基哌嗪環取代(如培氟沙星),
則脂溶性增加,腸道吸收增強,細胞的穿透性提高,半衰期延長。
在8位引進第二個氟原子,可進一步提高腸道吸收,延長半衰期(如洛美沙星等),
N-1修飾以環丙基團(環丙沙星)或噁嗪基團(氧氟沙星)可擴大抗菌譜,增強對衣原體、
支原體及分支桿菌(結核桿菌與麻風桿菌等)的抗菌活性,噁嗪環還可提高水溶性,使藥物在體內不被代謝,以原形經尿排泄。
(三)抗菌作用機制
喹諾酮類通過抑制DNA螺旋酶作用,阻礙DNA合成而導致細菌死亡。
大腸桿菌的DNA螺旋酶是四疊體結構的蛋白,由2個A亞單位與2個B亞單位組成,分子量分別為105kD與95kD(見圖42-1)。
細菌在合成DNA過程中,DNA螺旋酶的A亞單位將染色體DNA正超螺旋的一條單鏈(後鏈)切開,
接著B亞單位使DNA的前鏈後移,A亞單位再將切口封住,形成了負超螺旋。
根據實驗研究,氟喹諾酮類藥並不是直接與DNA螺旋酶結合,而是與DNA雙鏈中非配對鹼基結合,
抑制DNA抑螺旋酶的A亞單位,使DNA超螺旋結構不能封口,這樣DNA單鏈暴露,導致mRNA與蛋白合成失控,最後細菌死亡。
本類藥體外對DNA螺旋酶的半抑制濃度(IC50)與其對細菌的MIC呈一定的平行關係。
哺乳動物的細胞內也含有生物活性與細菌DNA螺旋酶相似的酶,稱為拓樸異構酶Ⅱ(topoisomerase
Ⅱ)。
氟喹諾酮類藥對人體細胞拓樸異構酶Ⅱ影響較小(見表42-1)。
從該表可見氧氟沙星與環丙沙星對動物細胞內拓樸異構酶Ⅱ的作用明顯比依諾沙星與萘啶酸小,IC50很高,
選擇指數很大。這可能是氧氟沙星與環丙沙星不良反應較少的原因。
喹諾酮類藥物-DNA結合抑制DNA螺旋酶活性的示意圖
圖42-1
喹諾酮類藥物-DNA結合抑制DNA螺旋酶活性的示意圖
圖中實心和斜線長方形示喹諾酮類藥物分子,A、B為DNA螺旋酶的A、B亞單位。
在DNA螺旋酶作用下,DNA雙鏈打開,而藥物分子嵌入雙鏈。與非配對鹼基結合,阻礙DNA雙鏈封口
(四)細菌耐藥機制
氟喹諾酮類藥物廣泛應用後,已出現細菌耐藥性。耐藥機理研究證實主要是染色體突變,不存在質體介導的耐藥性。
耐藥機制有二:
①細菌DNA螺旋酶的改變,與細菌高濃度耐藥有關;
②細菌細胞膜孔蛋白通道的改變或缺失與低濃度耐藥有關。
耐藥菌株DNA螺旋酶的活性改變主要由於gyrA基因突變所致。
(五)氟喹諾酮類藥理學共同特性
①抗菌譜廣,尤其對革蘭陰性桿菌包括綠膿桿菌在內有強大的殺菌作用,對金葡菌及產酶金葡菌也有良好抗菌作用;
某些品種對結核桿菌,支原體,衣原體及厭氧菌也有作用;
②細菌對本類藥與其他抗菌藥物間無交叉耐藥性;
③口服吸收良好,部分品種可靜脈給藥;體內分布廣,組織體液濃度高,可達有效抑菌或殺菌水平;
血漿半衰期相對較長,大多為3∼7小時以上。血漿蛋白結合率低(14%∼30%),多數經尿排泄,尿中濃度高;
④適用于敏感病原菌所致的呼吸道感染、尿路感染、前列腺炎,淋病及革蘭陰性桿菌所致各種感染,
骨、關節、皮膚軟組織感染;⑤不良反應少(5%∼10%),大多輕微,
常見的有噁心、嘔吐、食慾減退、皮疹、頭痛、眩暈。偶有抽搐精神症状,停藥可消退。
表42-1 喹諾酮類藥物對大腸桿菌和哺乳動物細胞DNA旋轉酶的選擇作用
藥物
IC50(mg/L)
選擇指數B/AA大腸桿菌KL-16DNA螺旋酶 B胎牛胸腺局部拓樸異構酶Ⅱ
氧氟沙星 0.76 1870
2461
環丙沙星 0.13 155
1192
依諾沙星 1.72 93
54
萘啶酸 23.0 385
17
二、各種喹諾酮類藥特點
吡哌酸(pipemidic acid,PPA)對革蘭陰性菌的抗菌作用較萘啶酸強,對革蘭陽性和部分綠膿桿菌有一定作用。
口服400mg後血濃度達不到治療效果,但尿中濃度高,可達900mg/L以上,主要用於治療尿路和腸道感染。
諾氟沙星(norfloxacin)又名氟哌酸,是第一個氟喹諾酮類藥,抗菌譜廣,抗菌作用強,對革蘭陽性和陰性菌包括綠膿桿菌均有良好抗菌活性,
明顯優於吡哌酸。口服吸收約35%∼45%;易受食物影響,空腹比飯後服藥的血濃度高2∼3倍,
血漿蛋白結合率為14%,體內分布廣,組織濃度高,藥物消除半衰期為3∼4小時。主要用於尿路及腸道感染。
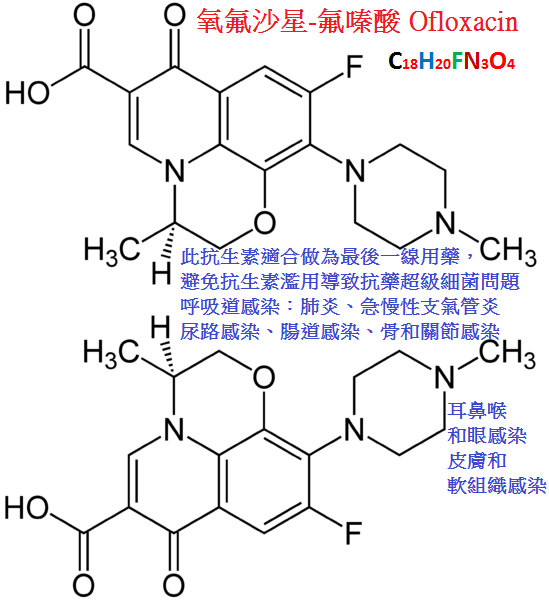
氧氟沙星(ofloxacin)又名氟嗪酸,抗菌活性強,對革蘭陽性菌(包括甲氧西林耐藥金葡菌,MRSA)革蘭陰性菌包括綠膿桿菌均有較強作用;
對肺炎支原體,奈瑟菌病,厭氧菌及結核桿菌也有一定活性。
對感染小鼠的保護效果明顯強於諾氟沙星、依諾沙星。
口服吸收快而完全,血藥濃度高而持久,血漿消除半衰期為5∼7小時,藥物體內分布廣,尤以痰中濃度較高,
70%∼90%藥物經腎排泄,48小時尿中藥物濃度仍可達到對敏感菌的殺菌水平,膽汁中藥物濃度約為血藥濃度的7倍左右。
依諾沙星(enoxacin)又名氟啶酸,抗菌譜和抗菌活性和諾氟沙星相似,對厭氧菌作用較差。
口服吸收好,不受食物影響,血藥濃度介於諾氟沙星與氧氟沙星之間,
口服後約50%∼65%經腎排泄,消除半衰期為3.3∼5.8小時。副作用以消化道反應為主,偶有中樞神經系統毒性反應。
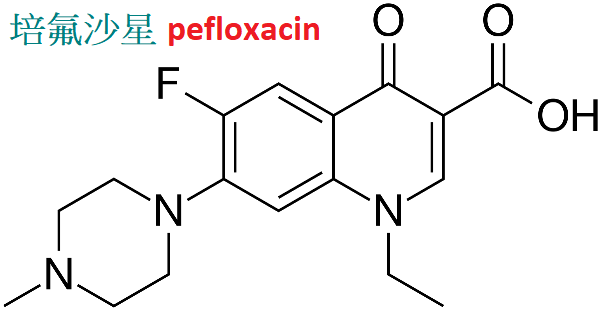
培氟沙星(pefloxacin)又名甲氟哌酸,抗菌譜廣與諾氟沙星相似,
抗菌活性略遜於諾氟沙星,對軍團菌及MRSA有效,對綠膿桿菌的作用不及環丙沙星。
服吸收好,生物利用度為90%∼100%。血藥濃度高而持久,半衰期可達10小時以上,體內分布廣泛,尚可通過炎症腦膜進入腦脊液。
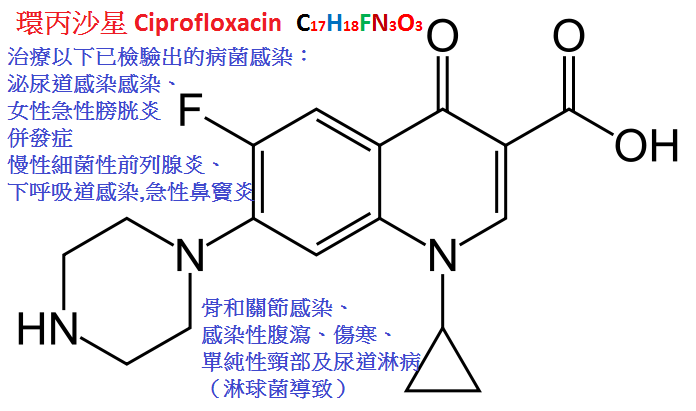
環丙沙星(ciprofloxacin)又名環丙氟哌酸,抗菌譜廣,
體外抗菌活性為目前在臨床應用喹諾酮類中最強,對耐藥綠膿桿菌,MRSA,產青黴素酶淋球菌、
產酶流感桿菌等均有良效,對肺炎軍團菌及彎麴菌亦有效,一些對氨基甙類、
第三代頭孢菌素等耐藥的革蘭陰性和陽性菌對本品仍然敏感。
口服後本品生物利用度為38%∼60%,血濃度較低,靜脈滴注可彌補此缺點。
半衰期為3.3∼5.8小時,藥物吸收後體內分布廣泛。
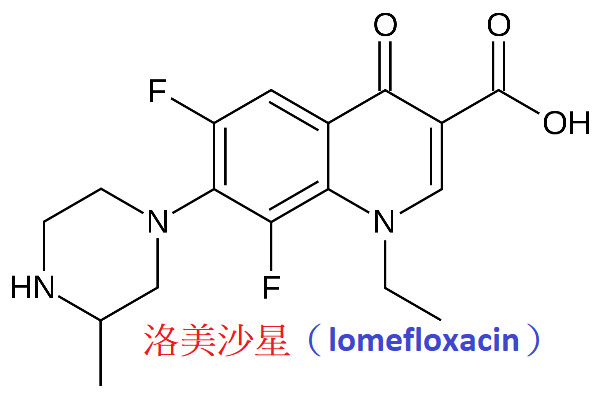
洛美沙星(lomefloxacin)抗菌譜廣,
體外抗菌作用與諾氟沙星、氧氟沙星、氟羅沙星相似,但比環丙沙星弱;
體內抗菌活性比諾氟沙星與氧氟沙星強,但不及氟羅沙星。
本品口服吸收好,生物利用度為85%,血藥濃度高而持久,半衰期約7小時,體內分布廣,藥物經腎排泄。
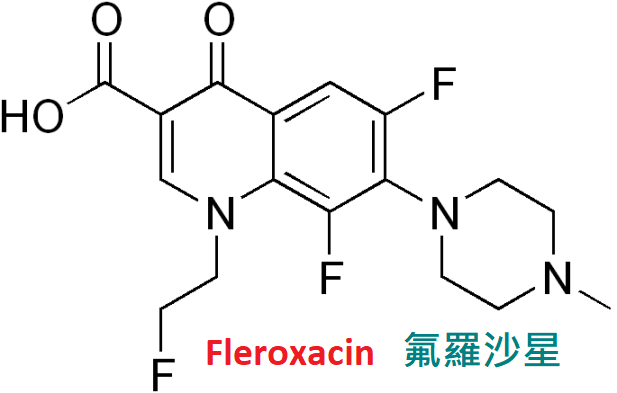
氟羅沙星(fleroxacin)又名多氟沙星,抗菌譜廣,
體外抗菌活性略遜於環丙沙星,但其體內抗菌活性強於現有各喹諾酮藥。
口服吸收好,生物利用度達99%。口服同劑量(400mg)的血藥濃度比環丙沙星高2∼3倍,
半衰期為9小時。體內分布廣,藥物經腎排泄,約為給藥量50%∼60%。
表42-2
幾種常用氟喹諾酮類藥的藥代動力學參數
藥物
單次口服劑量(mg) Cmax(mg/L)
t1/2kel(h)
絕對生物利用度(%)
Vd(L)
總清除率(L/h)
原藥尿中排泄率(%)
糞便排泄率(%)
諾氟沙星 400 1.58
3∼4
35∼45
>100 51.6
25∼30
28
培氟沙星 400 3.80
7.5∼11
90∼100 139
8.94
11
依諾沙星 400 3.70
3.3∼5.8
80∼89 175 21.0
52 18
氧氟沙星 400 5.60
5.0∼7.0
85∼95 120 12.84
70∼80 4
環丙沙星 500 2.56
3.3∼4.9
38∼60 307 39.12
29∼44 15
三、應用注意事項
1.對幼年動物可引起軟骨組織損害,故不宜用於妊娠期婦女和骨骼系統未發育完全的小兒。
藥物可分泌於乳汁,乳婦應用時應停止哺乳。
2.可引起中樞神經系統不良反應,不宜用於有中樞神經系統病史者,尤其是有癲癇病史的患者。
3.可抑制茶鹼類、咖啡因和口服抗凝血藥在肝中代謝,使上述藥物濃度升高引起不良反應。
產生上述相互作用最顯著者為依諾沙星、其次為環丙沙星與培氟沙星,氧氟沙星無明顯影響。
因此應避免與有相互作用的藥物合用,如有指征合用時,應對有關藥物進行必要的監測。
4.吸收,宜避免合用。
5.腎功能減退者應用主要經腎排的藥物如氧氟沙星和依諾沙星應減量。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}